

Multiple Bonds Between Metal Atoms / 08-Rhenium Compounds
.pdfRhenium Compounds 321
Walton
geometry (ρav = 24.4° versus 16.0° in 1,2,7-Re2Cl5(PMe3)3). Under other conditions Re2(µ- Cl)2Cl4(PMe3)4 can react to give mononuclear Re(III) or Re(IV) species.261
Chemistry similar to that described above for the PMe3 complexes of Re25+ and Re24+ has been developed with several other phosphines, including PMe2Ph. The relatively low cone angle phosphines PMe3 and PMe2Ph give very similar products. Depending on the choice of solvent, (Bu4N)2Re2Cl8 reacts with PMe2Ph at room temperature to form 1,3,6- and/or 1,2,7- Re2Cl5(PMe2Ph)3.180 The 1,2,7-isomer reacts with cobaltocene (in the presence of PMe2Ph) and with NOBF4 (in the presence of Bu4NCl) to afford 1,2,7,8-Re2Cl4(PMe2Ph)4 and 1,7- Re2Cl6(PMe2Ph)2, respectively,180 closely mirroring the behavior of 1,2,7-Re2Cl5(PMe3)3. The structures and electrochemical properties of the pairs of analogous PMe3 and PMe2Ph compounds are very similar (Tables 8.1, 8.2, 8.4 and 8.5). The reduction of 1,2,7-Re2Cl5(PMe2Ph)3 with KC8 in toluene/CH2Cl2, followed by the addition of an equivalent of Bu4NCl, gives (Bu4N)[1,2,7-Re2Cl5(PMe2Ph)3];262 this compound is similar structurally to its PMe3 analog (see Table 8.4). When the reduction of 1,2,7-Re2Cl5(PMe2Ph)3 by KC8 is carried out in the presence of PEt2H, the mixed phosphine complex 1,2,7,8-Re2Cl4(PMe2Ph)3(PEt2H) is formed.262 A similar compound, 1,2,7,8-Re2Cl4(PMe3)3(PEt2H), has been obtained, albeit in low yield and admixed with other products, when 1,2,7-Re2Cl5(PMe3)3 is reduced with cobaltocene and the reduced anion reacted with PEt2H.262 Both of these mixed-phosphine complexes have been structurally characterized (Table 8.4), and they differ only in their rotational geometries in the solid state, with a ρav value of 30.1° for the PMe2Ph complex and 3.0° for its PMe3 analog.262 The 1,2,7,8 ligand arrangement has (to date) been encountered only when the phosphine ligands have relatively small cone angles, i.e., PMe3, PMe2Ph and PEt2H.
As already mentioned in Section 8.5.2, salts of the [1,7-Re2Cl6(PR3)2]- anions can be generated by electrochemical means from their neutral Re26+ precursors,228 and also by their reaction with the one-electron reductant cobaltocene.178,190 In addition, the salts (Bu4N)Re2Cl6(PR3)2, where PR3 is PEt3, PPrn3 or PEt2Ph, have been isolated182,189 as the kinetic products in the reactions of these phosphines with (Bu4N)2Re2Cl8 in 1-propanol at room temperature; the PEt2Ph derivative was also obtained (although much more slowly) in benzene.189 The X-ray crystal structures of (Bu4N)Re2Cl6(PPrn3)2 and (Bu4N)Re2Cl6(PEt2Ph)2 have been determined (Table 8.4). In the case of the PPrn3 complex, both 1,7 and 1,6 isomeric forms exist in the solid state although in solution only one form (presumably the 1,7 isomer) is present.189 Both the 1,6- and 1,7-isomers of [Re2Cl6(PPrn3)2]- have partially staggered rotational geometries, while the PEt2Ph complex is eclipsed.189 The 1,3,6-Re2Cl5(PPrn3)3 complex was also isolated in different polymorphic forms during the course of these studies,182,189 each of which was characterized crystallographically (Table 8.4).
In the reaction between (Bu4N)2Re2Cl8 and PEt3 or PPrn3 in the non-polar solvent benzene,182 it is apparent that disproportionation occurs to give the dirhenium Re25+ products (Bu4N)Re2Cl6(PR3)2 or Re2Cl5(PR3)3 along with mononuclear trans-ReCl4(PR3)2. These disproportionations proceed by way of dirhenium(III) intermediates, which in the case of the PEt3 reaction has been identified as the unsymmetrical edge-shared bioctahedral complex (Bu4N)[(Et3P)2Cl2Re(µ-Cl)2ReCl3(PEt3)].182 In the case of these specific phosphines, the disproportionation can be represented as:
3Re26+ Α 2Re25+ + 2Re4+
Similar disproportionation behavior occurs with PEt2H183 and PCy2H181 when non-polar solvents are used, although the stoichiometries of the disproportionation processes are different in each case, and in turn differ from that given above for the PEt3 and PPrn3 reactions. In the reactions with these secondary phosphines, Re25+ and Re24+ complexes have been isolated and

322Multiple Bonds Between Metal Atoms Chapter 8
structurally characterized, namely, 1,2,7,8-Re2Cl4(PEt2H)4 and 1,3,6-Re2Cl5(PCy2H)3. Their structural and electrochemical properties are given in Tables 8.4 and 8.5.
Other examples of 1,3,6-Re2Cl5(PR3)3 are encountered with the phosphine-ester complexes Re2Cl5(Ph2PCH2CO2R)3, where R = Me or Et, which are prepared when (Bu4N)2Re2Cl8 and Ph2PCH2CO2H are reacted together in refluxing methanol or ethanol.263
It has become abundantly clear from several of these studies,181-183 that the reduction reactions that occur between (Bu4N)2Re2Cl8 and monodentate tertiary phosphines are greatly influenced by the reaction temperature, in some cases by the proportions of reactants, and (most importantly) by the choice of solvent. The solvent affects the reaction rate, the product solubility, and the reaction mechanism; alcohol solvents are implicated as being intimately involved in the reaction mechanism, while in non-polar solvents such as benzene, toluene, hexanes and dichloromethane a disproportionation mechanism seems to predominate (as demonstrated for PEt3, PPrn3, PEt2H and PCy2H).181-183
A re-investigation of the reactions between (Bu4N)2Re2I8 with PR3 ligands, which were originally carried out with ethanol or acetone as the reaction solvent and were shown to afford 1,3,6,8-Re2I4(PR3)4 complexes,25 has provided further insights into these systems.184 The use of either ethanol or benzene as the solvent at room temperature or below affords the isomers 1,3,6,8-Re2I4(PR3)4 (PR3 = PMe3, PEt3, PMe2Ph or PEt2Ph); the structures of several of these compounds were established by X-ray crystallography (Table 8.4). In the reaction of (Bu4N)2Re2I8 with PMe3 in benzene, the paramagnetic complex Re2(µ-I)2I4(PMe3)4 was isolated in high yield as a kinetic product.184 At room temperature it disproportionates to give some 1,3,6,8-Re2I4(PMe3)4, and when reduced with a two-fold excess of KC8 in toluene/dichloromethane it gives this same Re24+ complex along with a small amount of 1,3,6-Re2I5(PMe3)3 (Table 8.4). The analogous reaction of PEt3 with (Bu4N)2Re2I8 in benzene affords (Bu4N)[1,7- Re2I6(PEt3)2] as a kinetic product; its formation along with a Re(IV) species presumably occurs by a disproportionation mechanism.184
While the reductions of [Re2Cl8]2- and [Re2I8]2- by PR3 ligands are similar, there are several important differences.184 One of these is the much faster rate of reduction of [Re2I8]2- in ethanol, the solvent itself probably being involved in the reduction. While the same kinetic products are formed in benzene (i.e. Re2(µ-X)2X4(PMe3)4 and [Re2X6(PEt3)2]-) they are less stable for X = I. Also, the disproportionation products in this solvent are different, e.g., 1,3,6,8- Re2I4(PR3)4 versus 1,2,7-Re2Cl5(PR3)3. The different results of PR3 substitution on [Re2I8]2- versus [Re2Cl8]2- has been explained184 by the trans effect order Cl- > PR3 > I-.
In subsequent sections we will encounter additional aspects of the chemical reactivity of Re2X4(PR3)4 complexes. These will include the susceptibility of these Re>Re bonds to cleavage by /-acceptor ligands to afford mononuclear complexes (see Section 8.7). The use of Re2Cl4(PR3)4 as synthons for the preparation of the dirhenium octahydrides Re2H8(PR3)4 is also of note (see Section 8.8),264 as is the reaction of Re2Cl4(PEt3)4 with H2 in dichloromethane at 60 °C and 120 atm to give (Et4N)[Re2(µ-H)(µ-Cl)2Cl4(PEt3)2; the dirhenium(III) anion contains a short Re–Re distance (2.349(1) Å).265 An especially important reaction is the conversion of Re2X4(PR3)4 to complexes of the types Re2X4(LL)(PR3)2 and Re2X4(LL)2, where LL represents a bidentate (chelating or bridging) phosphine and/or arsine ligand.202 The latter reactions are discussed in the following sections.
Bidentate phosphines and arsines containing two or three bridgehead groups
For convenience, we will first deal with complexes that contain R2X(CH2)nYR2 ligands (R = alkyl or aryl; X = Y = P or As, and X = P when Y = As), or close analogs thereof, in which n = 2 or 3, and then with those cases in which there is only a single bridgehead group

Rhenium Compounds 323
Walton
present between the two donor atoms (i.e. n = 1). The first studies that were carried out involved the reactions of (Bu4N)2Re2X8 (X = Cl or Br) with the bidentate ligands 1,2-bis(di- phenylphosphino)ethane (dppe) and 1-diphenylphosphino-2-diphenylarsinoethane (arphos) in refluxing acetonitrile, from which the triply bonded dirhenium(II) compounds Re2Cl4(dppe)2, Re2Cl4(arphos)2 and Re2Br4(arphos)2 were isolated in low yield (< 12%).202 The reactions of (Bu4N)2Re2I8 with dppe and arphos in refluxing acetone for short periods lead to the iodide complexes Re2I4(dppe)2 and Re2I4(arphos)2 in much higher yields (60% and 30%, respectively) than those of their chloride and bromide analogs.25 The difference arises because in the chloride and bromide cases quite stable di-µ-halo bridged complexes, Re2(µ-X)2X4(LL)2, are also formed.200,202,209 A higher yield synthetic procedure utilized the reactions between Re2Cl4(PEt3)4 and dppe or arphos to afford Re2Cl4(dppe)2 and Re2Cl4(arphos)2.202 The arphos complex can also be prepared by reacting Re2Cl5(PEtPh2)3 with arphos in refluxing benzene. The mechanism for these reactions may well involve a disproportionation step similar to those that can occur in the reduction reactions of [Re2X8]2- with monodentate phosphines (vide supra).
The reversal of these substitution reactions has been accomplished in the case of the reaction between Re2Cl4(arphos)2 and PEt3 which gives 1,3,6,8-Re2Cl4(PEt3)4, thereby implying that a close structural similarity exists between the Re2X4(PR3)4 and Re2X4(LL)2 compounds. However, differences between the spectroscopic properties of Re2Cl4(LL)2 and 1,3,6,8-Re2Cl4(PR3)4 led to the proposal202 that although a trans-ReCl2P2 (or trans-ReCl2PAs) geometry is preserved in the Re2Cl4(LL)2 compounds, they have significantly different structures from Re2Cl4(PR3)4. Specifically, rather than the complexes containing an eclipsed rotational geometry and chelating LL ligands (i.e. as in structure 8.20), it was suggested202 that the bidentate ligands (LL) bridge the two metal atoms within the dimer thereby conferring a staggered rotational geometry (8.21). This was confirmed by a structure determination on Re2Cl4(dppe)2 (Fig. 8.18).266 The Re–Re distance of 2.244(1) Å is very similar to those in the Re24+ complexes Re2X4(PR3)4 (Table 8.4), which is to be expected since these complexes possess the same ligand sets, the same trans-Re2Cl2P2 geometry and the same Re–Re bond order. The electronic configuration μ2/4β2β*2 which is germane to Re2Cl4(PR3)4 imposes no rotational barrier, so with the displacement of PR3 by dppe (or arphos) the conformational preference of the chair-like Re2C2P2 rings that result (Fig. 8.18) apparently dominates, thereby ensuring the staggered conformation. In this structure the Cl–Re–Re–Cl and P–Re–Re–P torsional angles are 51° and 39°. Their deviations from 45° are doubtless attributable to the conformational demands of the six-membered rings.266 Subsequently, structures of the types represented by 8.20 and 8.21 were given the notation _-M2X4(LL)2 and `-M2X4(LL)2, respectively,267 a terminology that is still in common use, so that the aforementioned isomer is represented as `-Re2Cl4(dppe)2.
8.20 |
8.21 |
The structure determination of `-Re2Cl4(dppe)2 was an important milestone for several reasons, not the least of which is that it constituted the first example of its kind; many such complexes are now known, including several of dimolybdenum(II) and ditungsten(II). Also, this structure shows several characteristics that are often shared by other complexes with a structure

324Multiple Bonds Between Metal Atoms Chapter 8
like 8.21. Each crystal site is occupied by one or the other of two `-M2X4(LL)2 molecules, one orientation being far more prominent than the other.34 This sort of orientational disorder is related to that seen with the [Re2X8]2- anions (see Section 8.3), and is such that the M2 units of the principal and secondary molecules have the same midpoint and are approximately perpendicular. A consequence of the fused six-membered ring systems and the twisted geometry is that at each site, the principal and secondary isomers have opposite chiralities. Indeed, the chirality of such complexes has been of considerable interest, and this has led subsequently to the isolation of the first configurationally chiral dirhenium complex Re2Cl4(S,S-dppb)2, where S,S-dppb = S,S-2,3-bis(diphenylphosphino)butane, and its oxidized dication [Re2Cl4(S,S-dppb)2]2+ (vide infra).268,269 The CD spectrum of the quadruply bonded dication implies that it has the Ρ absolute configuration with a twist of less than 45°.268 The sort of twisting first encountered in the `-Re2X4(LL)2 complexes (structure 8.21), was later found in the related molybdenum and tungsten complexes of this stoichiometry (Chaps. 4 and 5).34 For example, the structure of the `-isomer of Mo2Br4(arphos)2270 has been determined, the mean torsional angle being c. 30°. Since quadruply bonded Mo24+ (and W24+) compounds have a μ2/4β2 configuration, the bond order will in these instances be reduced by rotation away from a full eclipsed structure unlike the situation with triply-bonded Re24+. The mean torsional angle of 30° is in accord with a bond order of c. 3.5 for `-Mo2Br4(arphos)2.270
Fig. 8.18. The structure of the Re2Cl4P4 skeleton in `-Re2Cl4(dppe)2 showing the staggered rotational geometry and the cis-decalin-like fusion of the two Re2C2P2 chair-like rings.
As an alternative to using (Bu4N)2Re2X8 (X = Cl, Br or I) and Re2X4(PEt3)4 (X = Cl or Br) as the starting materials for the synthesis of `-Re2X4(LL)2,25,202 the reactions of cis-Re2(O2CCH3)2X4(H2O)2 (X = Cl, Br or I) with dppe have been successfully employed to prepare `-Re2X4(dppe)2.271 In view of the general usefulness and availability of these carboxylate starting materials, this strategy has considerable merit. The compound `-Re2Cl4(dpae)2 (dpae = Ph2AsCH2CH2AsPh2) has been isolated as one of the products in the reaction between (Bu4N)2Re2Cl8 and dpae in refluxing n-butanol;272 an earlier report202 had described this reaction as yielding Re2Cl6(dpae)2. An X-ray structural analysis of `-Re2Cl4(dpae)2 crystals showed signs of Re2Cl6(dpae) as a minor component.272
Five years after the isolation of the first examples of `-isomers of Re2X4(LL)2, the complex Re2Cl4(dppp)2 (dppp = Ph2P(CH2)3PPh2) was obtained from the reaction of (Bu4N)2Re2Cl8 and dppp in refluxing acetonitrile.273 An X-ray crystal structure determination showed it to be the _-isomer and to have a structure as represented in 8.20, in which the two dppp ligands each chelate to a single metal atom and the conformation is eclipsed.273 More recently, this same compound was found to be the major product when toluene was used as the reaction solvent and the mixture refluxed for 4 days; the structure of a crystal of composition

Rhenium Compounds 325
Walton
_-Re2Cl4(dppp)2·4CH2Cl2 was reported (Table 8.4).189 A couple of years after the initial characterization of _-Re2Cl4(dppp)2,273 a second such compound, viz. _-Re2Cl4(dmpe)2 (dmpe = Me2P(CH2)2PMe2), was prepared from the reaction of (Bu4N)2Re2Cl8 with dmpe in metha- nol-conc HCl at room temperature. Its structure is similar to that of _-Re2Cl4(dppp)2, with a Re–Re distance of 2.266(1) Å (Table 8.4).274
It is now known that for most R2ECH2CH2ER2 ligands both _ and ` forms can be isolated and many have been structurally characterized. The first such pairs to be isolated and characterized were the _- and `-isomers of Re2X4(dppee)2 (X = Cl or Br; dppee = cis-Ph2PCH=CHPPh2)275 and the X-ray crystal structures of the chloro complexes determined (Table 8.4).276 The green _ forms are produced in low yields when (Bu4N)2Re2X8, or Re2Cl6(PBun3)2 in the case of the chloride system, are reacted with cis-dppee in refluxing methanol (acidified with conc HX) or ethanol; these conversions are accompanied by some cleavage of the dirhenium starting materials to give mononuclear complexes (see Section 8.7).275 The brown ` isomers are quite easily prepared through the use of Re2X4(PR3)4 (X = Cl or Br; R = Et or Prn) and their reaction with cis-dppee in refluxing benzene.275 This success in preparing both _ and ` isomers of a particular Re2X4(LL)2 complex eventually led to the successful preparation of _-Re2X4(dppe)2 (X = Cl or Br), using a procedure similar to that described for the analogous complexes containing cis-dppee.276 The crystal structures of two compounds that contain _-Re2Cl4(dppe)2 have been reported, namely, _-Re2Cl4(dppe)2·4C6H6 and _-Re2Cl4(dppe)2·dppe.277
The complexes `-Re2X4(dppee)2 are of special significance in that they constitute the first examples of compounds that contain the unsaturated ring system represented in 8.22.275,276 The resulting six-membered ring conformations are of a type well known for cyclohexene, namely, a flattened chair or half-chair.
8.22
The synthons (Bu4N)2Re2Cl8 and Re2X4(PR3)4 (X = Cl when PR3 = PEt3 and X = Br when PPrn3) have been used to prepare the isomeric pairs _- and `-Re2X4(depe)2, where depe is Et2PCH2CH2PEt2.275,278,279 The structures of the two isomers of the chloro complex show the usual features, with the _-form being eclipsed and having a slightly longer Re–Re bond than the staggered `-form.278,279
The compound _-Re2Cl4(dppbe)2 (dppbe = 1,2-bis(diphenylphosphino)benzene) has been prepared by the reaction between Re2Cl4(PPrn3)4 and dppbe in benzene or toluene.280 The electrochemical properties of _-Re2Cl4(dppbe)2 (see Table 8.5), as well as its low frequency infrared spectral properties, which closely resemble those of other _-Re2Cl4(LL)2 complexes,273,275,276 support the structural assignment. The `-isomer has not been isolated, which is to be expected in view of the more rigid nature of this ligand.
In addition to the extensive series of Re24+ complexes that contain R2E(CH2)nER2 ligands (n = 2 or 3) and which have been the subject of this section, a few compounds are known in which the paramagnetic Re25+ core is present. One of these is the salt [(δ5-C5H5)2Co][1,3-Re2Cl6(dppf)], that is formed by the cobaltocene reduction of Re2Cl6(dppf) and in which the anion has a structure of the type 1,3-Re2Cl6(PR3)2 (see 8.12).190 Another one is the salt (Bu4N)Re2Cl6(dppp),

326Multiple Bonds Between Metal Atoms Chapter 8
which is formed in the reaction of (Bu4N)2Re2Cl8 with 1,3-bis(diphenylphosphino)propane and is apparently an intermediate in the formation of _-Re2Cl4(dppp)2.189 It is a kinetic product of the reaction in toluene (at reflux) and acetonitrile (at room temperature), and an X-ray crystal structure determination shows it to have the expected 1,2-Re2Cl6(PR3)2 structure for the anion (Table 8.4).189 A third example, which like the dppp complex has a 1,2-structure for the anion, is formed when the tetrathiafulvalene ligand o-{Ph2P}2(CH3)2TTF (abbreviated o-P2) is reacted with (Bu4N)2Re2Cl8 in refluxing ethanol.281 The mixed-nuclearity salt [trans-ReCl2(o-P2)2][1,2- Re2Cl6(o-P2)] is obtained in high yield, the mononuclear cation being formed by non-reductive cleavage of some of the [Re2Cl8]2- anion. Reaction of this compound with cobaltocene reduces the cation to trans-ReCl2(o-P2) and forms (δ5-C5H5)[1,2-Re2Cl6(o-P2)].281
Like their analogs with monodentate phosphines, the dirhenium(II) compounds of the types _- and `-Re2X4(LL)2 exhibit two accessible one-electron oxidations. Electrochemical measurements on dichloromethane solutions of many of these complexes (see Table 8.5) have shown that the first process is reversible and that the potentials can be accessed by several one-electron oxidants.255,275,276,282 The kinetics of outer-sphere oxidation of `-Re2X4(dppee)2 (X = Cl or Br) by Co(III) has been studied.257 Several `-Re2X4(LL)2 complexes have been oxidized chemically to paramagnetic `-[Re2X4(LL)2]PF6 by NOPF6 in acetonitrile (X = Cl when LL = depe, dppe, dppe or arphos; X = Br when LL = dppee),275,282 and in a few instances the second oxidation has been achieved to give diamagnetic quadruply bonded `-[Re2Cl4(LL)2](PF6)2 (LL = depe, dppe or S,S-dppb).268,275 Note that `-[Re2Cl4(LL)2]+ cations do not appear to react with Cl-,282 in contrast to the reactivity of [Re2Cl4(PR3)4]+ to afford Re2Cl5(PR3)3 (vide supra). The monocationic species are EPR-active,275,282 and both monocations and dications retain the staggered rotational conformation of their parents as a result of the dominant conformational demands of the bridging phosphine ligands. Thus, the `-[Re2Cl4(LL)2]+ cations most probably possess a metal-metal bond order closer to 3, rather than that of 3.5, which would be expected in a system, like [Re2X4(PR3)4]+, where the rotational conformation is fully eclipsed. In a couple of instances, the related _ isomers have been oxidized successfully; both _-[Re2Cl4(dppee)2]PF6 and _-[Re2Cl4(dppp)2]PF6 have been obtained through the use of ferrocenium hexafluorophosphate as the oxidant.276
As a consequence of the shift of E1/2(ox) to more positive potentials for Re2X4(LL)2 relative to Re2X4(PR3)4 (Table 8.5), the reduction of Re2X4(LL)2 to the monoanions [Re2X4(LL)2]- becomes feasible. By the use of acetonitrile as solvent, in which [Re2Cl4(dppe)2]PF6 is soluble, and a hanging mercury drop electrode, an irreversible reduction in the potential range -1.6 to -1.7 V has been observed.282 This monoanion may possess a Re–Re bond order of 2.5. Also, in the cyclic voltammograms of _-Re2Cl4(dppe)2, _-Re2Br4(dppe)2 and _-Re2Cl4(dppbe)2 (recorded in 0.1 M Bu4NPF6-CH2Cl2),276,280 irreversible reductions have been measured between -1.4 and -1.6 V vs. Ag/AgCl.
While the complexes _-Re2X4(LL)2 and `-Re2X4(LL)2 have some limited chemical reactivity, such as the conversion of `-Re2Cl4(dppe)2 to Re2H8(dppe)2,264 and the reactions of `-Re2Cl4(dppe)2 and `-Re2Cl4(arphos)2 with CCl4 to regenerate the [Re2Cl8]2- anion,202 their oxidized congeners are much more reactive. This has been shown in studies detailing some of the reactions of `-[Re2X4(LL)2]PF6 (X = Cl when LL = depe, dppe or arphos; X = Br when LL = dppe or arphos).275,283 While their neutral precursors do not react with nitriles RCN (R = Me or Et), the salts `-[Re2X4(LL)2]PF6 react with these donors in the presence of TlPF6 to give paramagnetic `-[Re2X3(LL)2(NCR)](PF6)2. The latter complexes can be reduced to diamagnetic `-[Re2X3(LL)2(NCR)]PF6 with the use of LiBEt3H or cobaltocene as reductants.275,283 In all instances, spectroscopic and electrochemical data support the retention of the staggered rotational geometry of the parent species `-[Re2X4(LL)2]n+ (n = 0 or 1). The same is true of the

Rhenium Compounds 327
Walton
products formed from the reactions between `-[Re2X4(LL)2]PF6 (LL = dppe or arphos) and the isocyanide ligands RNC (R = Pri or But) in CH2Cl2.283 However, in these instances reduction to `-[Re2X3(LL)2(CNR)]PF6 occurs, although these Re24+ complexes can be oxidized to paramagnetic `-[Re2X3(LL)2(CNR)](PF6)2 with the use of NOPF6 as the oxidant.283
Bidentate and tridentate phosphines and arsines containing a single bridgehead group
The most famous of these is the ligand Ph2PCH2PPh2 (dppm). Although it can chelate, dppm shows a propensity for bridging two metal atoms. This is the situation in the case of Re2X4(µ-dppm)2 (X = Cl or Br), which were first prepared202 from the reactions between Re2X4(PPrn3)4 and dppm in benzene. Note that in this early report Re2Br4(µ-dppm)2 is referred to as `-[ReBr2(dppm)2]n. When Re2Cl4(PEt3)4 is used in place of Re2Cl4(PPrn3)4, the mixed phosphine complex Re2Cl4(µ-dppm)(PEt3)2 can be isolated (vide infra). Improved methods for obtaining Re2Cl4(µ-dppm)2 include the reaction of Re2Cl6(PPrn3)2 with dppm in methanol,203,274 although in diethyl ether the product is Re2Cl5(µ-dppm)2 (vide infra).203 However, perhaps the best method for preparing all three halide complexes of the type Re2X4(µ-dppm)2 (X = Cl, Br or I) involves the reactions between cis-Re2(O2CCH3)2X4L2 (L = H2O, 4-Mepy, DMF or DMSO) and dppm in refluxing ethanol.271 These reactions, which proceed via the intermediacy of Re2(O2CCH3)X4(µ-dppm)2 (at least in the case of X = Cl or Br), are part of an extensive chemistry involving mixed carboxylate-dppm complexes of dirhenium that will be discussed more fully a little later. The synthesis of Re2Cl4(µ-dppm)2 by this route can be simplified by use of a one-pot reaction between (Bu4N)2Re2Cl8, CH3CO2Na and dppm in ethanol.271
Several compounds analogous to Re2X4(µ-dppm)2 have been prepared by the use of procedures similar to those described above. By such means the compounds Re2X4(µ-dppa)2 (X = Cl or Br; dppa = Ph2PNHPPh2),203,284 Re2Cl4(µ-dcpm)2 (dcpm = Cy2PCH2PCy2),285 Re2Cl4(µ-cdpp)2 (cdpp = Ph2PC(CH2)2PPh2),286 Re2Cl4(µ-dppE)2 (dppE = 1,1-bis(diphenyl- phosphino)ethene)287 and Re2X4(µ-dpam)2 (dpam = Ph2AsCH2AsPh2)288 have been obtained. Another compound of the type Re2X4(µ-dppm)2 is the tetramethyl derivative Re2(CH3)4(µ-dppm)2 which is obtained by methylating the chloride complex with CH3Li.289 The compound Re2(NCBH3)4(µ-dppm)2(H2O)2·2THF has been prepared from the reaction between Re2Cl4(µ-dppm)2 and NaBH3CN in methanol,290 and similar reactions with Na[N(CN)2] and K[C(CN)3] give Re2[N(CN)2]4(µ-dppm)2 and Re2[C(CN)3]4(µ-dppm)2, respectively.291 The structure of the dicyanamide complex has been confirmed on a crystal of composition Re2[N(CN)2]4(µ-dppm)2(DMF)2·3DMF.291 The structure of this compound291 closely resembles that of Re2(NCBH3)4(µ-dppm)2(H2O)2·2THF290 and both compounds contain axially bound ligand molecules (Table 8.4). A different kind of behavior is encountered when Re2Cl4(µ-dppm)2 is reacted with (Bu4nN)CN in dichloromethane; cyanide for chloride substitution occurs plus coordination of additional cyanide to give the salt (Bu4N)2Re2(CN)6(µ-dppm)2, in which a long Re–Re single bond is present and two of the CN- ligands adopt an unusual δ2-(μ,/) bridging arrangement.292
Although the compound Re2Cl4(µ-dippm)2 (dippm = bis(di-iso-propylphosphino)methane) may well be formed in solution from the reaction between (Bu4N)2Re2Cl8 (or Re2Cl4(PMe3)4) and dippm, all attempts to isolate it have so far failed.293 It is in any event quite reactive and easily decomposes; in the presence of O2 the complex Re2O3Cl4(µ-dippm)2 is formed.293 In contrast to the ease of making Re2Cl4(µ-cdpp)2, the closely related complex Re2Cl4[µ-(2,2-dppp)]2 (2,2-dppp = Ph2PCMe2PPh2) cannot be obtained because the strong chelating tendency of this phosphine results in Re–Re bond cleavage to give trans-ReCl2(2,2-dppp)2.286
The first of the aforementioned Re2X4(µ-LL)2 compounds to be structurally characterized was Re2Cl4(µ-dppm)2.274 Subsequently, several of these compounds were characterized by X-ray

328Multiple Bonds Between Metal Atoms Chapter 8
crystallography, the results of which are summarized in Table 8.4. All show the same structural features, as typified in Fig. 8.19 by the structure of Re2Cl4(µ-dcpm)2, which clearly reveals the staggered rotational geometry.294 The extent of twisting can be defined in terms of the averages of the Cl–Re–Re–Cl and P–Re–Re–P torsion angles which are 43.6° and 39.0°, respectively, for the molecule shown in Fig. 8.19.
Fig. 8.19. The structure of Re2Cl4(µ-dcpm)2.
The group of complexes of the type Re2X4(µ-LL)2, have very similar spectroscopic and electrochemical properties. The spectroscopic characterizations have included electronic absorption spectroscopy and 1H and 31P{1H} NMR spectroscopy.285-290,294 Also, the low frequency infrared spectrum of Re2Cl4(dppm)2 shows two ι(Re–Cl) modes (335 s and 309 s cm-1) that mirror those found in the related spectrum of `-Re2Cl4(dppe)2 at 333 and 303 cm-1.202 The cyclic voltammetric properties of these complexes (Table 8.5), which resemble the behavior of the other dirhenium(II) complexes with bidentate phosphines and arsines, are discussed later in Section 8.5.4 when the redox chemistry of these complexes is considered.
An interesting and surprising result was obtained when attempts were made to prepare the bis(dimethylphosphino)methane complex Re2Cl4(µ-dmpm)2. The reactions of dmpm with (Bu4N)2Re2Cl8 in methanol and with Re2Cl4(PPrn3)4 in ethanol-toluene gave red crystalline Re2Cl4(µ-dmpm)3,295 and the bis-dmpm complex has so far defied all attempts to isolate it. The crystal structure of this complex was determined on different crystalline forms both of which are essentially the same (Table 8.4 and Fig. 8.20). The rotational conformation is staggered, and there is a two-fold axis passing through the methylene carbon atom of one dmpm ligand and the mid-point of the Re–Re bond. This defines the unique dmpm ligand (Fig. 8.20), which has a helical sense opposite to that of the other two, thereby making interconversions of the enantiomers by simple internal rotation about the Re–Re bond impossible. Not surprisingly, this results in a complex 1H NMR spectrum.295 The cyclic voltammetric properties of this complex show the presence of two accessible one-electron oxidations (Table 8.4).295 The related bromide derivative Re2Br4(µ-dmpm)3 has also been prepared and characterized (Table 8.4).296
With use of the tridentate phosphine bis[(diphenylphosphino)methyl]phenylphosphine, the 1:1 salts [Re2Cl3(dpmp)2]X (X = Cl or PF6) have been isolated upon reacting this ligand with (Bu4N)2Re2Cl8 in methanol.297 The structure of the cation in both salts is the same (8.23), with a staggered rotational geometry and the dpmp ligand doubly bridging the dirhenium unit.297
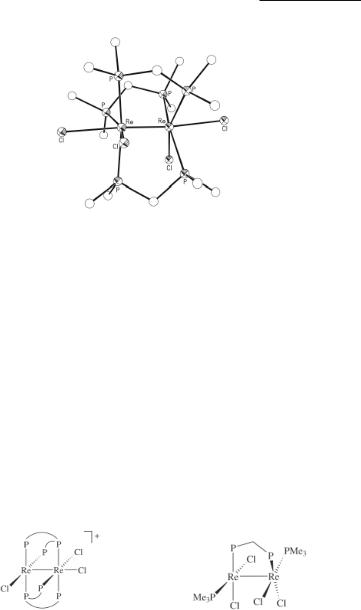
Rhenium Compounds 329
Walton
Fig. 8.20. The structure of the monoclinic form of Re2Cl4(µ-dmpm)3.
A variety of mixed-phosphine complexes that contain the dppm, dppa and dcpm ligands are also known. The first of these to be prepared was Re2Cl4(µ-dppm)(PEt3)2, which is obtained as a purple solid upon heating a mixture of Re2Cl4(PEt3)4 and dppm in benzene.202 Later, the PMe3 complexes Re2Cl4(µ-LL)(PMe3)2, where LL = dppm, dppa or dcpm, were obtained by a closely related procedure.244,285 The compound Re2Cl4(µ-dppm)(PMe3)2 has also been prepared by reacting Re2Cl4(µ-dppm)2 with an excess of PMe3, by heating a 1:1 mixture of Re2Cl4(PMe3)2 and Re2Cl4(µ-dppm)2 in 1-butanol and, quite unexpectedly, upon reacting the tripodal ligand HC(PPh2)3 with Re2Cl4(PMe3)4 in hot ethanol.298,299 In the direct reaction of Re2Cl4(µ-dppm)2 with PMe3, the 1:1 adduct Re2Cl4(µ-dppm)2(PMe3) is formed as an isolable intermediate.300 This complex, as well as its bromide analog and the corresponding phosphite derivatives Re2Cl4(dppm)2[P(OR)3] (R = Me, Et or Ph), have been characterized on the basis of their spectroscopic and electrochemical properties (Table 8.5).300 1H and 31P{1H}NMR spectroscopy has been used to show that Re2Cl4(µ-LL)(PMe3)2 possess structure 8.24, a conclusion that was confirmed by an X-ray crystal structure determination on Re2Cl4(µ-dppm)2(PMe3)2.285 These complexes, as well as the compound Re2Cl4(µ-dppa)(PMe2Ph)2,203 have redox properties that are typical of triply bonded Re24+ species (Table 8.5).244
8.23 |
8.24 |
The compounds Re2Cl4(µ-dppm)(PMe3)2 and Re2Cl4(µ-dppa)(PMe3)2 are converted to the ` isomers of Re2Cl4(dppm)(dppe), Re2Cl4(dppm)(arphos) and Re2Cl4(dppa)(dppe) upon their reaction with dppe or arphos in 1-butanol.298 Alternative synthetic strategies for Re2Cl4(dppm)(dppe) include the reaction of Re2Cl4(PMe3)4 with 2 equiv of dppm and 1 equiv of dppe, and the reaction of Re2Cl4(µ-dppm)2 with dppe.298 The spectroscopic and electrochemical properties (Table 8.5) of these mixed phosphine ligand complexes are in accord with their possessing a structure of the type shown in 8.21, with both bidentate ligands in a bridging transoid disposition to one another. This has been confirmed by a crystal structure determination of Re2Cl4(µ-dppm)(µ-dppe).298
330Multiple Bonds Between Metal Atoms Chapter 8
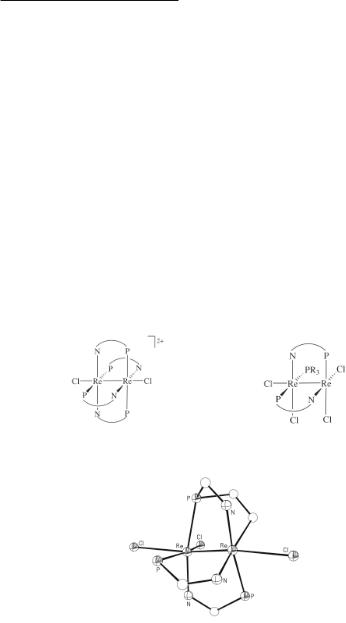
Several Re24+ compounds have been isolated that contain bridging ligands with N and P donor atoms rather than a pair of P atoms. A complex that is related to Re2X4(µ-dmpm)3 is Re2Cl4(µ-Ph2Ppy)3, which is formed from the reactions between (Bu4N)2Re2Cl8 or Re2Cl6(PBun3)2 and 2-(diphenylphosphino)pyridine in methanol.207,301 The Ph2Ppy ligand, like dppm, can bridge two metal atoms but in this instance the bridging atoms are N and P. While the complex Re2Cl4(µ-Ph2Ppy)3 has not been structurally characterized, it may have a structure related to that of Re2X4(µ-dmpm)3. However, this compound quite easily eliminates HCl to give the ortho-metalated complex Re2Cl3(µ-Ph2Ppy)2[(C6H5)(C6H4)Ppy],207,301 the first example of an ortho-metalation reaction occurring at a multiple bond in a molecule of the M2L8 type. The structure of Re2Cl3(µ-Ph2Ppy)2[(C6H5)(C6H4)Ppy] is shown in Fig. 8.21.207,301 Another product from this same reaction is the dirhenium(II) salt [Re2Cl2(µ-Ph2Ppy)4]Cl2. It can be metathesized with KPF6 to give [Re2Cl2(Ph2Ppy)4](PF6)2, a compound whose structure (represented in 8.25) has been determined by X-ray crystallography.207 This complex constitutes a rare example of a multiply bonded dimetal unit bridged by four neutral bridging ligands. In addition, the bis-Ph2Ppy complexes Re2Cl4(µ-Ph2Ppy)2(PR3) (R = Et or Bun) are the major products when Re2Cl6(PR3)2 are reacted with Ph2Ppy in acetone.207 They have the structure shown in 8.26, based upon an X-ray crystal structure determination of the PEt3 derivative. Note that the structures represented in 8.25 and 8.26 are both staggered with ρav torsion angles of 16.5° and 18.5°, respectively.207
8.25 |
8.26 |
Fig. 8.21. The structure of Re2Cl3(Ph2Ppy)2[(C6H5)(C6H4)Ppy] showing the central portion of the molecule with the phenyl and pyridyl rings omitted in order to emphasize the tridentate bonding mode of the orthometalated ligand.
While the electrochemical behavior of Re2Cl4(µ-Ph2Ppy)3, Re2Cl3(µ-Ph2Ppy)2- [(C6H5)(C6H4)Ppy] and Re2Cl4(µ-Ph2Ppy)2(PR3) shows a close resemblance to that of other triply bonded Re24+ complexes, the properties of [Re2Cl2(µ-Ph2Ppy)4](PF6)2 are different (Table 8.5).207 Apparently, there is increase in the effective positive charge at the dirhenium core in this dication relative to the other Ph2Ppy complexes, so that both the β* (the HOMO) and /* (the LUMO) orbitals fall in energy, thereby making oxidation (from β*) more difficult and reduction (through addition of electrons to /*) much easier.