

Multiple Bonds Between Metal Atoms / 08-Rhenium Compounds
.pdfRhenium Compounds 281
Walton
these dianions were interpreted in terms of N-bound thiocyanate and selenocyanate,63-65 and electronic absorption spectral measurements66 led to the conclusion that the β bond is weaker in [Re2(NCS)8]2- than [Re2Cl8]2- and [Re2Br8]2-. Much more recently, these interpretations have been substantiated by crystal structure determinations of the salts (Ph4As)2Re2(NCS)8(L)2 (L = (CH3)2CO or C5H5N).67 Both structures contain ‘solvent’ molecules (acetone or pyridine) that are weakly bound axially, the Re–O and Re–N distances being 2.56(1) Å and 2.54(1) Å respectively.67 This might be expected to result in a slight lengthening of the Re–Re bonds and, indeed, the measured distances of 2.270(1) Å and 2.296(1) Å, are longer than the corresponding distances in other [Re2X8]2- species (X = Cl, Br or I) (see Table 8.1). This bond lengthening is also encountered in the complex (Bu4N)2Re2(NCS)8(µ-dto)68 in which the weakly bridging 3,6-dithiaoctane ligand links [Re2(NCS)8]2- anions into infinite centrosymmetric chains (Fig. 8.3); the Re–S distance is 3.0072(8) Å.
Fig. 8.3. The structure of the infinite centrosymmetric chains present in (Bu4N)2Re2(NCS)8·dto.
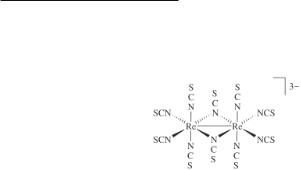
Whereas reflux in acidified methanol produces the octa(isothiocyanato)dirhenate(III) anion in the aforementioned reactions, the use of acetone as the reaction solvent produces solutions from which the red-brown rhenium(IV) complex (Bu4N)2Re(NCS)6 and a dark green material, originally formulated63 as (Bu4N)3Re2(NCS)10(CO)2, can be isolated. Several years later the true identity of this complex was established by X-ray crystallography.69 The structure of the anion is as shown in 8.1 and reveals at once the reason for the earlier misinterpretation of the infrared spectral data, namely, the presence of two N-bridging NCS ligands, a structural form of this ligand not previously documented. The Re–Re distance of 2.613(1) Å in this edge-shared bioctahedral complex implies the existence of a metal-metal bond, although its order is unknown and cannot be inferred from the magnitude of the Re–Re distance alone. The D2h symmetry of the Re2N10 core is consistent with the unpaired electron being delocalized equally over both metal atoms i.e. it is a (+3.5, +3.5) mixed-valence complex rather than being localized (+4, +3). However, it can safely be concluded that some degree of Re–Re multiple bonding exists in this species.70 Electrochemical studies have shown that [Re2(NCS)8]2- converts to [Re2(NCS)10]4- in the presence of free [NCS]- and that the latter species is readily oxidized to [Re2(NCS)10]3- (8.1) in the presence of oxygen,70 thereby explaining why the edge-shared bioctahedral [Re2(NCS)10]3- is so easily formed in the reaction between [Re2Cl8]2- and excess [NCS]-.63 It has also been established that at room temperature the one-electron oxidation of [Re2(NCS)8]2- to [Re2(NCS)8]- is followed by conversion of the latter to [Re2(NCS)10]2- via a [NCS]- scavenging mechanism involving the sacrifice of some unoxidized [Re2(NCS)8]2- (see also Section 8.5.2). More recently, the synthesis and crystal structures of a pair of salts that contain the [Re2(NCS)10]n- anions (n = 2 or 3) have been reported.71 Crystals of the bis(ethylenedithio)tetrathiafulvalene salts of compositions (BEDT-TTF)3Re2(NCS)10·2CH2Cl2 and (BEDT-TTF)2Re2(NCS)10·C6H5CN were prepared by electrooxidation techniques, and both complexes shown to have structures like
282Multiple Bonds Between Metal Atoms Chapter 8
8.1, with Re–Re distances of 2.602(1) Å for the [Re2(NCS)10]3- anion and 2.615(1) Å for the analogous [Re2(NCS)10]2- species.71
8.1
Although salts of the homoleptic methyl species [Re2(CH3)8]2- have been obtained, and one such compound structurally characterized (Table 8.1),72 they have not been prepared directly from the octahalodianions but rather from the dirhenium(III) carboxylates. Consequently, their chemistry is described in Section 8.4.2.
8.4.2 The dirhenium(III) carboxylates
The dirhenium(III) carboxylates Re2(O2CR)4X2, Re2(O2CR)3X3 and Re2(O2CR)2X4 (X = Cl, Br or I) not only have a very extensive chemistry in their own right but they have also occupied a crucial place in the development of the chemistry of the quadruple bond. A review of some of this chemistry, as seen from the perspective of Russian workers in the field, was published73 just prior to the publication of the second edition of Multiple Bonds Between Metal Atoms.10
Discovery, synthesis and structure
The recognition that rhenium(III) carboxylates of the type Re2(O2CR)4-xCl2+x (x = 0, 1 or 2) contain Re–Re quadruple bonds followed closely upon the heels of the structural characterization of the [Re2Cl8]2- anion and the original treatment of its bonding.7 Prior to that time, those literature reports that described low oxidation state rhenium carboxylates often failed to take into account the possibility of metal-metal bonding and in some cases explicitly precluded it. Accordingly, this early literature (pre-1965) is replete with erroneous conclusions concerning the nature of the materials that were purported to be formed.
The first report on low oxidation state rhenium carboxylates appears to be that published in 1958 by Kotel’nikova and Tronev13 who obtained a variety of products from the reactions between solutions containing “H2ReCl4·2H2O” and glacial acetic acid. The formulation of the products as derivatives of rhenium(II) (i.e. ReCl2·4CH3COOH, ReCl2·2CH3COOH·H2O, ReCl2·CH3COOH·H2O, ReCl2·CH3COOH and ReCl2·CH3COOH·C5H5N)13 is clearly incorrect, stemming in part from a failure to recognize that the solutions of “H2ReCl4·2H2O” contained, in reality, [Re2Cl8]2-. While further work in this period74,75 failed to establish the correct structural identity of these complexes, they were eventually assigned dimeric formulations.
Quite independently of the earlier Russian work, Taha and Wilkinson76 had, in their investigations into the reactions of rhenium(III) chloride (at the time of unknown structure) with mixtures of the lower monocarboxylic acids and the appropriate anhydride (when available), isolated crystalline orange products of stoichiometry [Re(O2CR)2Cl]n. The absence of air was essential for the reactions to proceed in this fashion (vide infra). Molecular weight measurements indicated that these complexes were dimeric and Taha and Wilkinson76 concluded that they most likely possessed the copper(II) acetate type of structure with terminally bound chlorines in the axial coordination sites. However, they further concluded76 that in spite of the diamagnetism of the complexes it was not necessary “to invoke metal-metal bonding to account for the

Rhenium Compounds 283
Walton
diamagnetism” and indeed explicitly reasoned against its existence. The lability of the chloride ligands was demonstrated76 by the reactions of [Re2(O2CC3H7)2Cl]2 with AgSCN and Ag2SO4. It is clear that the orange complexes formulated correctly by Taha and Wilkinson76 as the dirhenium(III) complex [Re(O2CCH3)2Cl]2 and incorrectly by Kotel’nikova and Vinogradova75 as (ReCl·2CH3COOH)2 are one and the same thing.
When the reactions between rhenium(III) chloride and the carboxylic acids were carried out in the presence of dry air or oxygen a mixture of purple [ReOCl(O2CR)2]2 and orange [ReO2(O2CR)2]2 was said76 to be produced. Both complexes were believed at the time to contain carboxylate and oxo-bridges and therefore to possess rhenium in an oxidation state higher than +3. However, later work by Lock and co-workers77,78 clarified the structural nature of these species.
While the reduction of KReO4 by hydrogen in hydrohalic acid (HCl or HBr)/carboxylic acid mixtures has continued to be used by Kotel’nikova and co-workers79,80 as a means of preparing the dirhenium(III) carboxylates, Re2(O2CR)4X2, the readily availability of high yield synthetic routes to (Bu4N)2Re2X8 (X = Cl, Br or I)22,29,54 provides a much more convenient synthetic procedure. The reaction of (Bu4N)2Re2Cl8 with an alkyl carboxylic acid, usually admixed with the appropriate anhydride and using oxygen and moisture free reaction conditions, is an excellent method for preparing Re2(O2CR)4Cl2.14,24,57,81
(Bu4N)2Re2Cl8 + 4RCO2H Α Re2(O2CR)4Cl2 + 4HCl + 2Bu4NCl
This strategy is readily applicable to the related bromide24,57,81 and iodide25 derivatives. Interestingly, in the reactions between [Re2X8]2- (X = Cl or Br) and monochloroacetic and monobromoacetic acids to prepare Re2(O2CCH2Cl)4Cl2 and Re2(O2CCH2Br)4Br2, halide ligand exchange also occurs (e.g., [Re2Cl8]2 + CH2BrCO2H Α Re2(O2CCH2Br)4Br2).82
In the case of the aryl carboxylic acids, the complexes are best prepared through carboxyl exchange utilizing the acetates.57,83
Re2(O2CCH3)4X2 + 2ArCO2H Α Re2(O2CAr)4X2 + 4CH3CO2H
As an alternative to starting with the [Re2Br8]2- and [Re2I8]2- anions, the following reaction84 of the pre-formed chloro-complexes Re2(O2CR)4Cl2 with liquid HBr or HI can be used:
Re2(O2CR)4Cl2 + 2HX Α Re2(O2CR)4X2 + 2HCl
Note that in the presence of an excess of HX and the appropriate Bu4NX salt, the latter reaction proceeds further to regenerate (Bu4N)2Re2X8 (see Section 8.3).25 The synthesis of the orange formate complex Re2(O2CH)4Cl2 has been accomplished through the reaction of (NH4)2Re2(O2CH)2Cl6 or Re2(O2CH)3Cl3 with formic acid at 70-80 °C in the presence of metallic zinc;85,86 the role of the zinc in these reactions is unclear. The infrared spectral properties of this complex have been compared with those of the analogous bromide although details for the preparation of Re2(O2CH)4Br2 were not described.85
The homoleptic acetate complex Re2(O2CCH3)6, which probably possesses the structure Re2(µ-O2CCH3)4(δ1-O2CCH3)2, has been isolated as the major product when the dirhenium octahydride complex Re2H8(PPh3)4 reacts with acetic acid/acetic anhydride mixtures in 1,2-di- chlorobenzene.87 This brown compound is very insoluble in most solvents but can be converted into Re2(O2CCH3)4Cl2 when reacted with gaseous hydrogen chloride in ethanol.
Recently, the first trifluoroacetate complex of dirhenium(III) was obtained by the solvothermal reaction of (Bu4N)2Re2Cl8 with CF3COOH/(CF3CO)2O mixtures.88 This affords the complex (Bu4N)Re2(O2CCF3)Cl6 (yield not reported), along with a reduced dirhenium by-product Re2(µ-Cl)2(CO)6. The reaction of (Bu4N)2Re2(O2CCF3)Cl6 with the organometallic carboxylic
284Multiple Bonds Between Metal Atoms Chapter 8
acid (CO)6Co2CHCCO2H gives the mixed-metal complex Re2[O2CCCHCo2(CO)6]4Cl2,88 which is similar structurally to other Re2(O2CR)4Cl2 carboxylates.
Several dirhenium(III) carboxylates of the type Re2(O2CR)4X2 have been fully characterized structurally78,81,83,88-91 and all are found to have the basic ‘paddlewheel’ structure represented in 8.2. These compounds, along with their Re–Re bond lengths, are listed in Table 8.1. From a historical perspective the most important structural determination was that carried out on the chloroform solvate of the dirhenium(III) benzoate, Re2(O2CPh)4Cl2·2CHCl3.89 This structure determination established that the Re–Re quadruple bond had indeed been retained upon substitution of the chloride ligands of the parent [Re2Cl8]2- anion by four bridging benzoate ligands. A further significant feature in the structure of the benzoate complex is the weakness of the axial Re–Cl bonds (r(Re–Cl) = 2.49 Å). In fact, the latter feature is common in the structures of all the dirhenium(III) carboxylate complexes of this type.
8.2
The weakness of the axial Re–Cl bonds in Re2(O2CR)4Cl2 is reflected in their substitutional lability; this fact has already mentioned in the case of the reactions of Re2(O2CR)4Cl2 with liquid HBr and HI to give Re2(O2CR)4X2 (X = Br or I).84 A further illustration is found in the recent report of the reactions of the pivalate complex Re2(O2CCMe3)4Cl2 with Na[M(CO)5CN] to form the linear µ-cyano bridged complexes Re2(O2CCMe3)4[µ-NCM(CO)5]2 (M = Cr, Mo or W).92 These complexes have been characterized primarily on the basis of their spectroscopic properties. Although detailed mechanistic studies of the halide substitution in complexes that contain the Re26+ core are quite rare, one such investigation by Webb and Espenson93 established that the reaction Re2(O2CC2H5)4Cl2 + Br- Α Re2(O2CC2H5)4ClBr + Cl- in acetonitrile proceeds by a two-step mechanism involving loss of Cl- prior to coordination of Br-. This reaction is subject to catalysis by trace amounts of such neutral donors as pyridine, DMF, urea, water, etc., an effect that has been ascribed93 to the nucleophilic character of the catalysts and their ability to stabilize the coordinatively unsaturated [Re2(O2CC2H5)4Cl]+ species.
The preceding discussion has focused on the carboxylate complexes of the type Re2(O2CR)4X2, which represent the maximum extent to which substitution of the halide ligands of the parent [Re2X8]2- anions may occur. Bearing in mind the description by Kotel’nikova et al13,74,75 of materials that were said to be (ReCl2·CH3COOH·H2O)2 and Re2Cl3·(CH3COOH)3·H2O, the existence of Re2(O2CR)2X4 and Re2(O2CR)3X3, representing intermediate degrees of substitution of [Re2X8]2-, seemed likely. Indeed, Kotel’nikova and co-workers later published several reports79,80,94,95 that provided details of the experimental conditions necessary for the conversion
of KReO4 and K2ReX6 to Re2(O2CR)2X4(H2O)2 (R = CH3, C2H5, (CH3)2CH, (CH3)3C or C6H5; X = Cl or Br) through the high pressure hydrogen reduction of these reagents in mixtures of HX
and RCO2H. Several mixed halide derivatives of the type Re2(O2CCH3)2Cl4-xBrxL2 (L = DMF, DMSO, or DMA) were obtained serendipitously as by-products during the autoclave synthesis

Rhenium Compounds 285
Walton
of Re2(O2CCH3)2Br4L2.96 It appears that the presence of chloride in these products arose from impurities that had been adsorbed on the wall of the autoclave from previous experiments that had involved an HCl-containing reaction mixture.
An alternative and very convenient synthesis of Re2(O2CCH3)2X4(H2O)2 (X = Cl or Br) and Re2(O2CC2H5)2Cl4(H2O)2 that has been developed involves the reaction of (Bu4N)2Re2X8 with acetic or propionic anhydride and 48% aqueous HBF4.97,98 The blue trichloroacetate complex Re2(O2CCCl3)2Cl4 is produced97 when (Bu4N)2Re2Cl8 is added to molten trichloroacetic acid.
When the aforementioned hydrated acetato complexes Re2(O2CR)2X4(H2O)2 (X = Cl or Br) are reacted with neutral donor ligands (L) such as pyridine, 4-methylpyridine, dimethylacetamide, DMF, DMSO and Ph3PO, the coordinated water molecules are displaced to give Re2(O2CR)2X4L2.79,98,99 The relationship between the Raman active Re–Re stretching frequency and the donor properties of the monodentate neutral ligands in complexes of the type Re2(O2CCH3)2Cl4L2 has been examined.100 The pyridine complex Re2(O2CCH3)2Cl4(py)2, which is formed upon treatment of an aqueous solution of Re2(O2CCH3)2Cl4(H2O)2 with pyridine, must be identical with the material described by Kotel’nikova and Vinogradova74 as “(ReCl2·CH3COOH·C5H5N)2”. Also, a close similarity in the spectroscopic properties of this group of complexes (including the hydrates) implies79 that they are closely related structurally. Crystal structure determinations on representative members of this series (vide infra) have shown that in all instances except one, namely the benzoate containing [Re2(O2CPh)2Cl6]2- anion, as present in the salt [ReCl2(dpcp)2]Re2(O2CPh)2Cl6,101 there is a cis-arrangement of bridging carboxylate groups and the ligands L are axially bound; accordingly, they are usually represented as cis-Re2(O2CR)2X4L2. This is also true in the case of the chloride complex (Bu4N)Re2(O2CCH3)2Cl5, which consists of individual cis-Re2(O2CCH3)2Cl4 units linked into infinite chains by bridging axial chloride ligands.101 While Re2(O2CPh)2Cl4(THF)2·THF has the usual cis structure, the bis-chloride adduct [Re2(O2CPh)2Cl6]2-, possesses a transoid arrangement of carboxylate ligands.101
More recently, it has been reported that Re2(O2CR)2X4(H2O)2 reacts with the symmetrical linker ligands pyrazine, 4,4'-bipyridine and methylenebis(diphenylphosphine oxide) to form insoluble polymeric complexes of the type [Re2(O2CCH3)2Cl4(LL)]n in which the cis structure of the Re2(O2CCH3)2Cl4 unit is preserved.102 In addition, the non-polymeric pyrazine complex [Re2(O2CCH3)2Cl4(pyz)]2(µ-pyz) has been isolated in which both terminally bound and bridging pyrazine ligands are present.102
Several formato complexes that are derived from Re2(O2CH)2Cl4 are known. The entry to this chemistry is through (NH4)2Re2(O2CH)2Cl6, a complex that is formed by the reaction of (NH4)2Re2Cl8 with formic acid.85,103 The corresponding Cs+ salt has also been prepared and shows very similar infrared spectral properties to (NH4)2Re2(O2CH)2Cl6.85 Treatment of this complex with DMF or diphenylformamide in formic acid gives cis-Re2(O2CH)2Cl4(DMF)2 and cis-Re2(O2CH)2Cl4(DPF)2.85,104,105 On the other hand, with triethylamine as the base, the bluegreen crystalline salt [(C2H5)3NH]Re2(O2CH)3Cl4·HCO2H is formed.106 The crystal structure of this salt suggests that it is best considered as a derivative of Re2(O2CH)2Cl4. The pyridine adduct Re2(O2CH)2Cl4(py)2 is formed from (NH4)2Re2Cl8·2H2O in the presence of formic acid and pyridine.105 The pyrolysis of Re2(O2CH)2Cl4L2 (L = DPF, DMF or py) has been reported to form ReCl(CO)5 and ReO2 as the main decomposition products.105 While the formate complex Re2(O2CH)2Br4(DMF)2 has been described as precipitating when a solution of K2Re2Br8·2H2O is treated with formic acid and DMF,85 it remains poorly characterized.
Since the tetrakis(formate) derivative Re2(O2CH)4Cl2 (vide supra) can be prepared from (NH4)2Re2(O2CH)2Cl6, it is logical that, with the careful manipulation of the reaction conditions, Re2(O2CH)3Cl3 can also be prepared from this same starting material upon its reac-

286Multiple Bonds Between Metal Atoms Chapter 8
tion with formic acid.85,86,107 A few other examples of carboxylates of the type Re2(O2CR)3X3 are known. Thus, while Re2(O2CCH3)2X4L2 complexes are converted into Re2(O2CCH3)4X2 upon prolonged reflux with glacial acetic acid,79,97 much milder and more controlled reactions have been used to convert Re2(O2CCH3)2X4(H2O)297 and Re2(O2CCH3)2X479 (X = Cl or Br) into Re2(O2CCH3)3X3. However, as discussed below, an additional method for preparing Re2(O2CR)3X3 compounds is through the thermal decomposition of Re2(O2CR)4X2.
Thermal studies have shown95,108,109 that the axial ligands L of Re2(O2CCH3)2X4L2 can be lost on heating, although it is apparent that this can be a complex process. The most thoroughly studied systems are the hydrates; the volatile anhydrous acetates Re2(O2CCH3)2X4 are formed following loss of H2O.95 X-ray structure determinations on anhydrous Re2(O2CCH3)2X4 have shown110-112 that a trans arrangement of acetate ligands is present, thereby establishing that the loss of the axially bound ligand molecules is accompanied by a cis Α trans isomerization. This isomerization process is reversed upon the re-addition of the axial ligand.113 The thermal decomposition of several carboxylates of the type Re2(O2CR)4X2 (X = Cl or Br; R = CH3, C2H5, (CH3)2CH, (CH3)3C and C6H5) has also been found to yield Re2(O2CR)2X4 compounds under a flow of inert gas (Ar or N2),112 but the stoichiometry of the reactions are quite complex and, in the case of Re2(O2CC2H5)4Cl2, appreciable quantities of Re2(O2CC2H5)3Cl3 are also said to be produced.112 The latter observation is in accord with earlier results from the thermal decomposition of the pivalate complex Re2(O2CCMe3)4Cl2 in vacuo. At a temperature of 240 °C the major product is pink Re2(O2CCMe3)3Cl3, whereas at 260 °C green Re2(O2CCMe3)2(HO2CCMe3)Cl4 is produced.114 Resublimation of the latter complex at 160 °C in a sealed tube leads to loss of the molecule of pivalic acid and the formation of green crystals of Re2(O2CCMe3)2Cl4.114
There are other reactions that afford some of these same carboxylates, by less obvious pathways than the ones that start from [Re2X8]2-, i.e., the starting materials do not already contain a Re–Re quadruple bond. Thus, at the beginning of this section (8.4.2) we described the reactions of trinuclear rhenium(III) chloride with alkyl carboxylic acids. The complexes [Re(O2CR)2Cl]2 which had been isolated in this fashion by Taha and Wilkinson76 are the same orange colored species Re2(O2CR)4Cl2 that were later prepared in a more logical fashion14 directly from [Re2Cl8]2-. Another example is rhenium(IV) chloride, [`-ReCl4] , which can be converted into Re2(O2CCH3)4Cl2 upon reflux with acetic acid, and also to dark blue Re2(O2CCH3)2Cl4(H2O)2.115 However, perhaps the most interesting system, is trans-ReOX3(PPh3)2 (X = Cl or Br). The reactions of these mononuclear complexes are rather complicated and the products that are formed depend critically upon the reaction conditions.116 Mixtures of red trans-ReCl4(PPh3)2, purple Re2OCl3(O2CR)2(PPh3)2 (the latter originally mis-formulated as lacking the oxygen) and/or dark green Re2OCl5(O2CR)(PPh3)2 are obtained upon heating trans-ReOCl3(PPh3)2 with carboxylic acids in boiling toluene. Structural studies117,118 on Re2OCl5(O2CC2H5)(PPh3)2 and Re2OCl3(O2CC2H5)2(PPh3)2 revealed the correct formula for the latter and showed that these complexes are carboxylate bridged edge-sharing bioctahedral dirhenium compounds that contain Re–Re bonds. Although the Re–Re bond distances are quite short (2.51-2.52 Å) it is not clear what these values imply in terms of the metal-metal bond orders. Upon prolonged hearing of Re2OCl3(O2CR)2(PPh3)2 with more carboxylic acid the quadruply-bonded complexes Re2(O2CR)4Cl2 are produced.116 Alternatively, the latter may be prepared in fairly high yield by the direct reaction of trans-ReOCl3(PPh3)2 with the refluxing acid anhydride. Accordingly, Re2OCl5(O2CR)(PPh3)2 and Re2OCl3(O2CR)2(PPh3)2 represent intermediate stages of reduction in the conversion of ReOCl3(PPh3)2 to Re2(O2CR)4Cl2. From the reactions between trans- ReOBr3(PPh3)2 and the carboxylic acids or their anhydrides, the related bromide complexes Re2(O2CR)4Br2 can be prepared.116

Rhenium Compounds 287
Walton
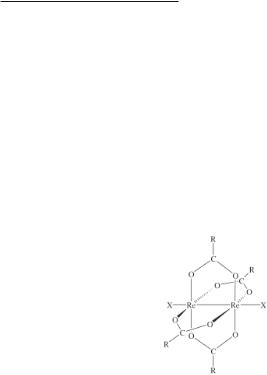
Around the time of the structure report on Re2(O2CPh)4Cl2,89 which was the first for a quadruply-bonded compound of the type Re2(O2CR)4Cl2, Koz’min et al119 described preliminary details of a structure determination on the carboxylate complex they represented as ReCl2·CH3COO(H)·2H2O. Three years later the full structure report appeared,120 the complex continuing to be represented incorrectly as containing rhenium(II) and acetic acid i.e. Re2Cl4[CH3COO(H)]2·2H2O. The Re–Re bond length (2.224(5) Å) is consistent with a quadruple bond, and there are axially bound water molecules (r(Re–O) = 2.50 Å) and a cis-arrangement of bridging acetate groups. This same type of structure (8.3) has been found for all complexes of the type Re2(O2CR)2X4L2 that have been structurally characterized (R = H when L = DPF; R = CH3 when L = H2O, DMSO or DMF),104,120-122 including (NH4)2Re2(O2CH)2Cl6 in which L = Cl-. The axial Re–Cl distances in the latter complex are very long (2.71 Å) relative to the equatorial Re–Cl bond lengths (2.31 Å).123 The polymeric ligand-bridged complexes [Re2(O2CCH3)2Cl4(µ-LL)]n, where LL = pyz, 4,4'-bpy or dppmO2, as well as [Re2(O2CCH3)2Cl4(pyz)]2(µ-pyz), also contain the 8.3 unit; all these compounds have been crystallographically characterized (Table 8.1).102 Polymeric complexes have also been isolated with isonicotinamide (INA) and nicotinamide as the axial ligands.124 In these cases, polymerization arises from strong intermolecular hydrogen-bond interactions. A further variation of 8.3 is seen in the structure of the formate complex [(C2H5)3NH]Re2(O2CH)3Cl4·HCO2H, in which cis-Re2(O2CH)2Cl4 units are linked by axially bridging formate ligands to form polymeric chains.106 The structures that are based on 8.3 are listed in Table 8.1.
As was mentioned earlier in this section, ligand loss from cis-Re2(O2CR)2X4L2 produces Re2(O2CR)2X4, which have been shown to possess the symmetric trans structure 8.4. In the structures of the acetate complexes Re2(O2CCH3)2X4, the Re–Re bond distances are 2.2084(3) Å for X = Cl and 2.216(3) Å for X = Br; the neighboring dinuclear units are linked via weak Re–X···Re bridges (2.887(1) Å for X = Cl and c. 3.09 Å for X = Br).110-112 These structures are very similar to those reported earlier for Re2(O2CPh)2I458 and Re2(O2CCMe3)2Cl4,114 which in turn resemble the centrosymmetric structures of the amidinate complexes Re2[(PhN)2CPh]2Cl4 and Re2[(PhN)2CCH3]2Cl4 that are discussed in Section 8.4.3. The blue chloro complex Re2(O2CCH3)2Cl4 preserves its identity in the gas phase as shown by mass spectral measurements.111
8.3 |
8.4 |
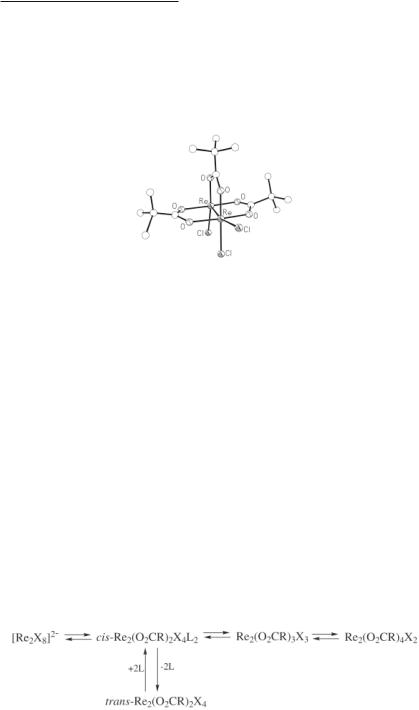
Turning now to the 3:3 complexes Re2(O2CR)3X3, the pivalate complex Re2(O2CCMe3)3Cl3 can be considered to have the prototype structure (Fig. 8.4). It represents a situation that is intermediate between Re2(O2CR)4Cl2 and Re2(O2CR)2Cl4. Parallel chains of [Re2(O2CCMe3)3Cl2]+ units are linked by bridging Cl- ligands that are shared between the axial positions of successive [Re2(O2CCMe3)3Cl2]+ ions in the chains.114 A similar structure was reported around the same time for the formate complex Re2(O2CH)3Cl3.107
With the basic structural information available for the three main groups of dirhenium(III) carboxylates, it is now appropriate to return to the question of the structures of complexes for-
288Multiple Bonds Between Metal Atoms Chapter 8
mulated by Taha and Wilkinson76 as [ReOCl(O2CR)]2 and [ReO2(O2CR)]2 that they obtained upon refluxing Re3Cl9 with carboxylic acids in the presence of oxygen. Lock and co-workers77,78 have shown by structural studies on the two butyrate derivatives that these complexes are in reality [Re2(O2CR)3Cl2]ReO4 and [Re2(O2CR)4](ReO4)2, respectively, so they correspond to known structural types, i.e. Re2(O2CCMe3)3Cl3 and Re2(O2CCMe3)4Cl2, with perrhenate substituted for axial halide.
Fig. 8.4. The structure of Re2(O2CCMe3)3Cl3 showing the formula unit.
In addition to studies of the thermal decomposition of various formato complexes of dirhenium(III)86,105and1HNMRspectralstudiesoftheisomerizationbetweentrans-Re2(O2CR)2X4 and cis-Re2(O2CR)2X4L2,113 an assortment of carboxylates, namely, Re2(O2CCH3)4Br2, trans- Re2(O2CCH3)2X4 (X = Cl or Br), cis-Re2(O2CCH3)2X4L2 (L = py, DMSO, etc.), Re2(O2CH)2Cl4L2 (L = DMF or DMSO), and (NH4)2Re2(O2CH)2Cl6 have been studied by 35Cl or 81Br NQR spectroscopy and the results compared with data for salts containing the [Re2Cl8]2- and [Re2Br8]2- anions.125
Reactions in which the Re26+ core is preserved
In this section, we consider those non-redox reactions of the dirhenium(III) carboxylates in which the dirhenium unit remains intact. Most redox reactions of these carboxylates, and reactions in which the Re–Re quadruple bond is cleaved, are discussed in Sections 8.5 and 8.7, respectively.
On the basis of the previous discussions of the synthetic procedures that have been used to obtain the dirhenium(III) carboxylates, the interconversions shown below are well documented and need not be considered in any further detail. Suffice it to say that the reversibility of these reactions serves to illustrate dramatically the stability of the Re–Re quadruple bond. Note that these same reactions are also encountered in the case of the various dirhenium(III) formate complexes.85 Thus, (NH4)2Re2(O2CH)2Cl6, Re2(O2CH)3Cl3 and Re2(O2CH)4X2 (X = Cl or Br) all react with NH4Cl in conc HCl to give (NH4)2Re2Cl8·2H2O.85
Behavior related to that mentioned above is encountered in the reactions of Re2(O2CCH3)4Cl2 with the gaseous hydrogen halides (HX).126-128 In all instances, these reactions, when carried out at 300-350 °C, lead to complete displacement of the acetate groups and the formation of the trinuclear rhenium(III) halides Re3X9. In the case of the reaction with HCl(g), the bright blue solid that is formed as an intermediate126 has been shown to be trans-Re2(O2CCH3)2Cl4.111

Rhenium Compounds 289
Walton
The synthetic utility of the dirhenium(III) carboxylates is further shown by their usefulness in providing a convenient entry to various alkyl derivatives that contain the Re26+ core. The interaction of the benzoate Re2(O2CPh)4Cl2 with methyllithium in diethyl ether produces72 Li2Re2(CH3)8·2Et2O, a diamagnetic red crystalline complex. It is airand water-sensitive but thermally stable. Addition of tetramethylethylenediamine or 1,10-phenanthroline to ether solutions of Li2Re2(CH3)8·2Et2O yields pyrophoric Li2Re2(CH3)8·tmed and Li2Re2(CH3)8·phen. The etherate, Li2Re2(CH3)8·2Et2O, can also be produced by the reaction of rhenium(V) chloride with methyllithium. This is an especially significant reaction since it constitutes a relatively rare example of the formation of a Re–Re quadruple bond from a mononuclear starting material without the use of bridging ligands. The crystal structure of Li2Re2(CH3)8·2Et2O has been determined72 and reveals the short Re–Re bond (Table 8.1) and eclipsed configuration that are so characteristic of the presence of a Re–Re quadruple bond.
The reactions of Li2Re2(CH3)8·2Et2O with various monodentate tertiary phosphines gives Re2(CH3)6(PR3)2 in good yield,129 a reaction course analogous to that in which the [Re2X8]2- anions are converted to Re2X6(PR3)2 (Section 8.4.4). Mixed alkyl-carboxylato complexes can be obtained starting from either Re2(O2CCH3)4Cl2 or [Re2(CH3)8]2-. The treatment of Re2(O2CCH3)4Cl2 with R2Mg reagents in diethyl ether produces red crystalline Re2(O2CCH3)2R4, where R = CH2Si(CH3)3, CH2C(CH3)3, CH2C(CH3)2Ph or CH2Ph.130 The trimethylsilylmethyl and neopentyl derivatives are quite air stable. When Re2(O2CCH3)4Cl2 reacts with three equivalents of bis-2-methoxyphenylmagnesium, the diamagnetic dark green dirhenium(III) complex Re2(2-CH3OC6H4)6 is produced.131 This complex is of unknown structure although it probably retains a Re–Re quadruple bond; its 1H and 13C NMR spectra are consistent131 with aryl groups in two different environments.
The addition of glacial acetic acid and acetic anhydride to Li2Re2(CH3)8·2Et2O gives the bright red air-stable complex Re2(O2CCH3)4(CH3)2. Its crystal structure has been determined (Fig. 8.5), revealing132 that it does not have the Re2(O2CR)4Cl2 type structure. Two acetate groups bridge the quadruply-bonded pair of rhenium atoms while a chelate acetate and terminal methyl group are bound to each metal atom.132 The oxygen atoms of the chelating acetate ligands that occupy the axial coordination sites of the dinuclear complex (along the Re–Re axis) are, as expected, weakly bound (2.46 Å versus 2.02-2.12 Å for the equatorial Re–O bonds).132 It has been suggested,133 that the treatment of Re2(O2CH)4Br2 with Et3Al leads to Re2(O2CH)4Et2 and perhaps Re2(O2CH)4H2, prior to cleavage of the Re–Re bond to give mononuclear products. However, neither complex has been isolated and definitively characterized.133
Fig. 8.5. The structure of Re2(O2CCH3)4(CH3)2.
Treatment of Re2(O2CCH3)4(CH3)2 with chlorine in dichloromethane and with methanol gives130 purple-mauve powders of stoichiometry [Re(O2CCH3)Cl(CH3)]n and [Re(O2CCH3)- (OCH3)(CH3)]n, respectively. These two materials probably bear a close structural relationship to one another. The chloro-derivative is soluble in dimethylsulfoxide from which air stable crystals of Re2(O2CCH3)2Cl2(CH3)2·DMSO have been grown. The basic structure of this complex132

290Multiple Bonds Between Metal Atoms Chapter 8
shows a close resemblance to that of trans-Re2(O2CCH3)2Cl4, with a DMSO ligand occupying only one of its two available axial coordination sites. A rather surprising feature in the structures of the methyl derivatives Re2(O2CCH3)2Cl2(CH3)2·DMSO and Re2(O2CCH3)4(CH3)2 is the shortness of the Re–Re bonds (Table 8.1) even though both complexes contain axial ligands.132 These appear to be examples of molecules for which the presence of axial ligands does not lead to an obvious weakening of the Re–Re bond.
The substitution of all four carboxylate groups in Re2(O2CR)4X2 (especially when R = CH3 and X = Cl) by other monoanionic bridging ligands has often been used to prepare compounds of the type Re2(µ-bridge)4X2. This procedure often constitutes a convenient alternative to the use of [Re2X8]2-. These reactions are more appropriately discussed in Section 8.4.3, along with those of the [Re2X8]2- anions with monoanionic bidentate ligands. However, note the possibility of obtaining complexes in which there are mixed sets of carboxylate and other monoanionic bridging ligands. Such an example is the complex Re2(O2CEt)2(9-EtA)2Cl2 in which there are cis-pairs of bridging propionate and the bridging anion of the DNA nucleobase adenine, the latter being bound through its N1 and N6 positions.134
The substitution chemistry of Re2(O2CCH3)2X4L2 (X = Cl or Br; L = H2O or 4-methyl- pyridine) has also proved synthetically useful,135 although many of the reactions of these compounds lead to Re25+ and Re24+ species (see Section 8.5.4). Examples of non-redox substitution chemistry include reactions with 2-hydroxypyridine and with 2-methyl-6-hydroxypyridine in THF or acetone which afford Re2(hp)2Cl4·Hhp·THF and Re2(mhp)2X4·Hmhp·S (S = THF or (CH3)2CO), respectively (for further details see Section 8.4.3).136 While Re2(O2CCH3)2Cl4(H2O)2 reacts with an excess of Hhp in acetonitrile or ethanol to give Re2(hp)4Cl2, the Hmhp ligand reacts with Re2(O2CCH3)2X4(H2O)2 (X = Cl or Br) in a different fashion when nitrile solvents R'CN (R' = CH3 or C2H5) are used. The latter reactions afford products of the type Re2(O2CR')(mhp)2X3, in which the bridging carboxylate ligands is formed by hydrolysis of the R'CN solvent.136
A variety of interesting and synthetically useful reactions that involve dirhenium(III) carboxylates and tertiary phosphine ligands have been examined. Several of these lead to Re25+ and Re24+ products and so are discussed in Section 8.5.4, while others will be considered here. Most of these involve products in which all of the carboxylato ligands have been replaced but in a couple of cases a carboxylate ligand is retained. In the case of the reaction between Re2(O2CPh)4Cl2 and PMe3 in ethanol in the presence of traces of air (or a small amount of H2O2) an unusual centrosymmetric ‘dimer of dimers’ complex [(PMe3)3ClRe(µ-O2CPh)Re(O)]2(µ-O)2 is obtained, the structure of which is shown in Fig. 8.6.137 The rhenium centers in each dirhenium unit have distinctly different coordination environments i.e. P3ClORe–ReO4, and it has been suggested that this unit can be considered formally as Re(IV)–Re(II) or Re(V)–Re(I). The Re–Re bond distance of 2.3396(8) Å is consistent with a Re–Re multiple bond. Other examples that give rise to this general type of intramolecular disproportionation had been encountered previously. Thus the reaction of Re2(O2CCH3)4Cl2 with Cy2PCH2PCy2 produces both ReOCl(δ2-dcpm)2 and trans-Re2(µ-O2CCH3)2Cl2(µ-dcpm)2 (see Section 8.5.4), the mononuclear Re(III) product subsequently converting to O3ReReCl(δ2-dcpm)2 (8.5) upon exposure to O2.138 The Re–Re distance in 8.5 is 2.5398(6) Å and therefore signifies a bond order less than four. A formal description of this compound as a Re(VI)–Re(I) system seems reasonable.138 In an even earlier report it was shown that the reactions of the bis-acetato complex cis- Re2(O2CCH3)2Cl4(H2O)2 with Me2PCH2PMe2 gives a compound that is related structurally to 8.5, namely, O3ReReCl2(dmpm)2.139 This reaction, like the aforementioned one that gives 8.5, might proceed through a mononuclear intermediate. In the dmpm compound, the two metal centers have coordination numbers of four and seven as shown in 8.6, rather than four and six as