

Multiple Bonds Between Metal Atoms / 04-Molybdenum Compounds
.pdfMolybdenum Compounds 119
Cotton
upon its reaction with a methanol solution of Me4NCl followed by the addition of a solution of dppm in dimethoxyethane.369 The treatment of the methyl derivative Mo2(CH3)4(PMe3)4 itself prepared from Mo2(O2CCH3)4, with conc. HCl in methanol produces the blue chloride Mo2Cl4(PMe3)4.226
The only compounds containing X = F are Mo2F4(PMe3)4 and Mo2F4(PMe2Ph)4. The anion Mo2F84− is unknown, and the preparation of these compounds391 was accomplished by the reaction:
where “Olah’s reagent” (OR) is a 70% solution of HF in pyridine. The compounds are relatively unstable, especially toward visible light, but are well characterized by 19F and 31P NMR, and the structure of Mo2F4(PMe3)4 was confirmed by X-ray crystallography (Mo–Mo = 2.110(5) Å).
The reactions of acetone solutions of (Bu4N)Mo2Br6 with pyridine, PEt3, PPrn3, dppe or arphos result in reduction of this bromo-anion and the formation of Mo2Br4L4 and Mo2Br4(LL)2 compounds.340 This starting material is of uncertain nuclearity but it could well be tetranuclear. Indeed, other reactions are known in which tetranuclear molybdenum clusters degrade to dinuclear species. Thus, the 2,2'-bipyridyl complex Mo2I4(bpy)2 is formed upon prolonged reflux of an acetonitrile solution of (Bu4N)2Mo4I11, with bpy.340 Also, the `-MoX2 phases (X = Cl, Br or I),331,337 which are believed to contain tetranuclear clusters of molybdenum atoms333 react with an excess of monodentate PR3 to afford Mo2X4(PR3)4. The reactions of the salt (Bu4N)2Mo2Br6 (formally the one-electron reduced congener of (Bu4N)Mo2Br6)340,342 with PEt3, PEt2Ph, dppe and (Ph2PCH2CH2)2PPh (bdpp) are said343 to give complexes of the type (Bu4N)[Mo2Br5L2], (Bu4N)[Mo2Br5L4] or Mo2Br4L4, depending upon the choice of reaction conditions. However, the complexes that are formulated as Mo2Br4(PEt3)4, Mo2Br4(PEt2Ph)4 and Mo2Br4(dppe)2 are described343 as being orange in color, quite different from the colors that are normally associated with authentic samples of these complexes (blue-purple for the PEt3 and PEt2Ph complexes, green for _-Mo2Br4(dppe)2 and red-brown for `-Mo2Br4(dppe)2).331,368,377 Accordingly, some question exists as to the true identity of these particular products.343
A curious route to complexes of the type Mo2X4(PR3)4 is the reaction of molybdenum atoms with oxalyl chloride to give a material that upon extraction into THF and treatment with PEt3 affords Mo2Cl4(PEt3)4.431 A route to Mo2Br4(PMe3)4 involves the decomposition of the triply bonded dimolybdenum(III) complex Mo2Br2(=CHSiMe3)2(PMe3)4 in hydrocarbon solvents:432
3Mo2Br2(=CHSiMe3)2(PMe3)4 Α Mo2Br4(PMe3)4 + 2MoBr(>CSiMe3)(PMe3)4 + 2Me4Si + Me3SiCH=CHSiMe3 + other product(s)
There are also the very slow reactions between the trihalides MoX3 and tertiary phosphines in refluxing ethanol or toluene to give Mo2X4(PR3)4 (X = Cl, Br or I; R = Me, Et or Prn).345,433 Since the solid-state structures of MoCl3434 and MoBr3435 are based on face-sharing MoX6 octahedra with adjacent metal atoms drawn together in pairs (Mo–Mo = 2.76 Å in MoCl3 and 2.92 Å in MoBr3), the formation of Mo2X4(PR3)4 may involve the cleavage of the halide bridges and retention and enhancement of the Mo–Mo interactions of the trihalides. The dimethylamine and trimethylamine complexes Mo2X4(HNMe2)4 (X = Cl or Br) and Mo2Cl4(NMe3)4 have been prepared from the trihalides.344-346 In the case of the bromide/dimethylamine system, these results corrected an earlier formulation of the product as the solvolyzed molybdenum(III) complex MoBr2(NMe2)·NHMe2.436 Both of the dimethylamine complexes are readily convertible to Mo2X4(PPrn3)4, thereby supporting345 this structural formulation.
In contrast to the relatively sluggish reactivity of the trihalides themselves, the THF complexes MoCl3(THF)321 or Mo2Cl6(THF)3352,351 provide much more convenient routes. Also, the

120Multiple Bonds Between Metal Atoms Chapter 4
comproportionation reaction between MoI3(PMe3)3 and Mo(CO)6 in refluxing toluene gives Mo2I4(PMe3)4.362 Other examples are known where a higher oxidation state mononuclear molybdenum complex is reduced in a ‘one-pot’ reaction to give Mo2X4(PR3)4 compounds. When an excess of hydrochloric acid is added to the hydride MoH4(PMePh2)4, monomeric MoCl3(PMePh2)3 is formed, but when THF is used as the reaction solvent and the HCl:MoH4(PMePh2)4 stoichiometric ratio is adjusted to 2:1, then the green complex Mo2Cl4(PMePh2)4 can be isolated.359 This reaction represents formally the reductive elimination of hydrogen and the coupling of pairs of low oxidation state coordinatively unsaturated molybdenum monomers. Attempts to purify this green compound were thwarted359 by its conversion to a more stable blue isomer. A similar result was obtained by Luck and Morris440,439 who prepared this same complex in its green and blue forms by a comproportionation reaction involving the reaction of Mo(δ6-PhPMePh)(PMePh2)3 with MoCl4(THF)2. The complex Mo2Cl4(PMe2Ph)4 was prepared by a similar procedure, as was Mo2Cl4(PEt2Ph)4 although in an impure form.439 Another example of a mononuclear to dinuclear transformation is that reported by Sharp and Schrock,437 who found that the sodium amalgam reduction of a THF solution of MoCl4 and PBun3 gave Mo2Cl4(PBun3)4 via the intermediacy of MoCl4(PBun3)2.
In addition to the methods outlined in Table 4.7, an additional but little-used strategy is halide exchange, which has been used438 to convert Mo2Cl4(dppm), to its bromo and iodo analogs by reaction with NaX in acetone. However, a problem with this method is ensuring that complete halide replacement occurs.416 Phosphine exchange can also be used, as in the conversion of Mo2Cl4(PMePh2)4 to Mo2Cl4(PMe3)4.439 In a few instances the synthesis of compounds of the type `-Mo2X4(LL)2 is best approached by allowing the preformed _-isomer to isomerize to the more thermodynamically stable `-form in solution, e.g. `-Mo2Cl4(dptpe)2 and `-Mo2Cl4(R-dppp)2 whose preparations have not been reported by any other means.383 In a related context, other solution reactions of note include the slow conversions (ligand redistribution reactions) of Mo2(O2CCH3)Cl3(PMe3)3 in THF to a mixture of Mo2Cl4(PMe3)4 and Mo2(O2CCH3)4,127 and of Mo2(O2CCH3)2Cl2(dppm)2 to Mo2Cl4(dppm)2 in several solvents.123,190
Most molecules of the Mo2X4L4 type are the 1,3,6,8 isomers,356,358,362,363,366 presumably because the usually larger L ligands best avoid one another that way. However molecules of the type Mo2Cl4(Rpy)4, where Rpy may be 4-Mepy, 4-Butpy or 3,5-Me2py, are remarkable in their capacity to present themselves with a variety of rotation angles about the Mo–Mo bond in different crystals.351 Some are close to having D2h symmetry (1,3,5,7), some close to D2d (1,3,6,8) and a few are well in between with only D2 symmetry. In the D2d and D2h structures there is essentially full β overlap and it must be small differences in intramolecular nonbonded forces that decide the outcome. For the D2 structures, where much of the β overlap has to be lost, various nonbonded interactions evidently dominate. A subsequent study403 of these molecules by spectroscopy in solution and DFT calculations (B3LYP with large basis sets) led to two principal conclusions: (1) The D2h (1,3,5,7) conformation, though frequently found in crystals, is the least stable in solution or the vapor phase. (2) The relative stabilities of the D2d (1,3,6,8) and D2 conformations are both solvent-dependent and temperature-dependent.
The vast majority of Mo2X4L4 and Mo2L4(LL)4 compounds have monoor diphosphines as neutral ligands, but before proceeding to these the Mo2X4(amine)4 compounds will be discussed. In fact, the first Mo2X4L4 compound ever reported436 (1962) was then thought to be MoBr2(NMe2)·NHMe2, rather than, as now recognized, Mo2Br4(NHMe2)4. Later investigations established the existence of Mo2Cl4(NHMe2)4 and Mo2Cl4(NMe3)4 as well.344-346
The three Mo2X4(amine)4 compounds just mentioned were prepared from MoIII starting materials. Yields were low and the way in which reduction of some of the molybdenum occurs remains obscure. More recently, the preparation and chemical reactions of Mo2X4(amine)4 com-

Molybdenum Compounds 121
Cotton
pounds were further studied.339 While spontaneous reduction of Mo2Cl6(THF)3 in the presence of NHEt2 does occur to give low yields of Mo2Cl4(NHEt2)4, the use of Na/Hg as a reductant allows efficient preparation:
Mo2Cl6(THF)3 + 2Na/Hg + 4NHEt2 Α Mo2Cl4(NHEt2)4
By similar reactions, Mo2Cl4(amine)4 compounds with the primary amines NH2Et, NH2Prn, NH2But and NH2Cy have been prepared in almost quantitative yield and characterized.352
It is a general characteristic of the Mo2X4(amine)4 compounds that the amines may be displaced by phosphines. This point was studied in detail364 for Mo2Cl4(NHEt2)4, where displacement is facile, and it was shown that the Mo2X4(PR3)4 compounds with PR3 = PMe3, PMe2Ph, PHEt2, δ1-Me2PCH2PMe2 and δ1-Me2PCH2CH2PMe2 are obtained smoothly. The latter two are quite novel in that the normally bidentate dmpm and dmpe ligands are attached to metal atoms by only one phosphorus atom, with the other one dangling, as shown in Fig. 4.15 for Mo2Cl4(δ1-dmpm)4. On heating, this compound expels two dmpm molecules to form the previously known ß-Mo2Cl4(dmpm)2:
Mo2Cl4(δ1-dmpm)4 Α Mo2Cl4(δ2,µ-dmpm)2 + 2dmpm
The Mo2Cl4(δ1-dmpe)4 compound also decomposed on heating, but in a complex way that led to an unidentified solid. When Mo2Cl4(NHEt2)4 reacted with dppa and dppm the products were the conventional Mo2Cl4(LL)2 molecules.
Fig. 4.15. The structure of 1,3,6,8-Mo2Cl4(δ1-dmpm)4.
For the Mo2Cl4(NH2R)4 compounds, replacement of the NH2R by phosphines is less facile than for NHEt2; replacement proceeds only halfway at ambient temperature and heating is necessary to go all the way to an Mo2Cl4(PR3)4 product. Also, back reaction occurs. It was possible to show in detail the stepwise nature of these reactions.405 The general results are summarized in Fig. 4.16, although the details vary with the particular amine and phosphine used, depending particularly on the basicity of the latter. The overall pattern displays a “stereochemical hysteresis,” in that the forward and reverse paths are not identical. The reason for this is the large difference in the trans influence of phosphines and amines; the former is far greater. Thus, the action of PR3 on Mo2Cl4(PR3)(NH2R)3 leads to isomer (3) of the Mo2Cl4(PR3)2(NH2R)2 intermediate, whereas the action of NH2R on Mo2Cl4(PR3)3(NH2R) leads to isomer (4) because the preference is always to replace a ligand opposite to a PR3 group rather than one opposite to a NH2R group.
The blue mixed-ligand complex Mo2Cl4(PPh3)2(CH3OH)2 was isolated365 during attempts to prepare Mo2Cl4(PPh3)4 through reaction of (NH4)5[Mo2Cl9]·H2O with PPh3 in methanol. It

122Multiple Bonds Between Metal Atoms Chapter 4
is the centrosymmetric isomer with a 1,3,5,7 distribution of neutral ligands. Upon dissolution in benzene it is converted to a brown complex of stoichiometry [MoCl2(PPh3)]n. Reaction of the latter material with the trialkyl phosphines PEt3 or PBun3 in benzene at 25 °C converts it to diamagnetic brownish-yellow complexes that proved to be tetranuclear Mo4Cl8(PR3)4.365 The chemistry of these and other tetranuclear molybdenum complexes is dealt with in Section 4.5.6.
An interesting subtlety addressed in the X-ray structure determination of Mo2Cl4(PMePh2)4,363 concerns the relationship between the green and blue forms of this complex. These two forms had been encountered in prior synthetic studies359,439,440 and their electronic absorption and 31P NMR spectra were found359 to be essentially the same. However, there are some differences in their low frequency infrared spectra359 and their electrochemical properties are quite different (see below).439 Based on the crystal structure of the blue form, which is the 1,3,6,8- isomer, it has been suggested363 that the difference lies in the orientation of the PMePh2 ligands about the Mo–P bonds. The blue form is the form with the least degree of repulsive contact (S4 symmetry).363
Fig. 4.16. The interconversion of Mo2Cl4(PR3)4 and Mo2Cl4(NRH2)4 compounds, showing the dual pathway. For simpicity the neutral ligands are represented by P and N.

Molybdenum Compounds 123
Cotton
Studies of electronic absorption spectra (particularly the β Α β* transi-
tion),102,331,347,350,353,358,361,366,404,439 low frequency infrared spectroscopy (ι(Mo–X)).102,331,346,347,350,354
Raman spectroscopy (ι(Mo–Mo)),102,347,350,366,404,441 and 31P NMR spectroscopy248,362,363,439 have been used to identify compounds as containing Mo2X4L4 molecules. The presence of a β Α β* transition close to 600 nm, two infrared-active ι(Mo–X) modes, a Raman-active ι(Mo–Mo) mode at c. 350 cm−1, and a singlet in the 31P{1H} spectrum are particularly characteristic. Excited state spectra have been studied in considerable detail for Mo2X4(PMe3)4,442-444 Mo2Cl4(PBun3)4,445 and Mo2Cl4(NCCH3)4,445 and the gas-phase PE spectrum of Mo2Cl4(PMe3)4 has been recorded and interpreted in terms of the μ2/4β2 configuration.446 The latter spectroscopic studies442-446 are discussed in more detail in Chapter 16.
We turn now to the Mo2X4(LL)2 class of compounds, in which the bidentate ligands, LL, are almost always diphosphines. The structures that have been established by X-ray crystallography are listed in Table 4.9.
The structure of the complexes that contain a single atom between the two donor atoms of the LL ligands are relatively simple as shown in Fig. 4.17 in the case of the monoclinic form of `-Mo2Cl4(dmpm)20.367 This molecule, which possesses a rigorously eclipsed rotation geometry and bridging dmpm ligands has the phosphorus atoms in the 1,3,5,7 arrangement. Most of the molecules that contain diphosphinomethane ligands do not have precisely eclipsed structures. As Table 4.9 shows, twist angles of 30° or more are observed. The dppm complexes Mo2X4(dppm)2 (X = Cl, Br or I) show a similar structure371 although Mo2I4(dppm)2, when grown from CH2Cl2/CH3OH, crystallizes with two independent molecules in the unit cell, one of which is centrosymmetric with an average torsional angle (ρ) of zero, while the other possesses no crystallographically imposed symmetry and has an average ρ of 17°.416 This result demonstrates clearly that crystal packing forces can play an important role in determining the exact rotational geometry. The complex Mo2Cl4(tdpm)2 (tdpm = (Ph2P)3CH) can be considered as containing a modified dppm ligand.370 It resembles the aforementioned structures but with an uncomplexed Ph2P unit replacing one of the hydrogen atoms of the bridgehead CH2 group. Also, the molecule assumes a partially staggered conformation (ρ = 20[3]°), presumably because of the steric bulkiness of the extra PPh2 group.370
Fig. 4.17. The structure of the monoclinic form of `-Mo2Cl4(dmpm)2.
When the LL ligands have the two donor atoms separated by two (or even three) carbon atoms they may be attached to the Mo2X4 unit in either of two ways, as shown in 4.25 for biphosphines.

124Multiple Bonds Between Metal Atoms Chapter 4
4.25
In the ` isomers it should be noted that the fusion of two 6-membered rings along the Mo– Mo bond results, in every case but one (see below), in a non-zero torsion angle about this bond. Apart from any influence that packing forces may have, the angle of rotation reflects a balance between conformational preferences of the rings and the retention of β-bonding. It has been estimated420 that with a torsion angle of 30° about half of the β-bond strength is retained.
The actual occurrence of _ and ` isomers was first recognized for molybdenum compounds (although analogous ones were already known for Re2X4(LL)2 molecules) when Mo2Cl4(LL)2 compounds containing dppe, arphos and dpae ligands were made and structurally characterized.340,368
Following this early work a profusion of both _ and ` isomers of Mo2X4(LL)2 compounds have been made and characterized structurally, spectroscopically and in other ways. The vast majority of the structures that have been determined crystallographically are those of ` isomers because these are generally more stable than their _ analogs. Some _ isomers have been observed to isomerize to their ` analogs, and in many cases the _ isomer has not been observed. The twist angles in ` isomers range from ~0° to ~70°, but the majority are in the range of 20° to 40°. The only case in which both _ and ` isomers of the same stoichiometry have been characterized crystallographically is Mo2Cl4(dppe)2.378,417 There is also one case, `-Mo2I4(dppe)2·0.67CH2Cl2, in which two independent molecules are present, one with ] = 27.9° and the other 0°. The latter is shown in Fig. 4.18.
Fig. 4.18. The structure of the eclipsed rotomer (ρ = 0) of `-Mo2I4(dppe)2.
For several of these structures, different kinds of structural disorder have been encountered. In the cases of `'-Mo2Cl4(dmpe)2373 and `-Mo2Cl4(depe)2374 there is a disorder of the Cl and phosphine ligands that imparts a higher crystal symmetry than that of the individual molecules. Specifically, there is a twofold axis coincident with the Mo–Mo axis, and two other

Molybdenum Compounds 125
Cotton
twofold axes perpendicular to the Mo–Mo axis. For `-Mo2Cl4(dmpe)2 there is also another form of disorder that is found with some other `-Mo2X4(LL)2 molecules listed in Table 4.9. This is an orientation disorder involving primary (or major) and secondary (or minor) orientations of the Mo2 unit that are essentially orthogonal. This disorder is quite commonly encountered with `-Mo2X4(LL)2 compounds and is of the same kind as that commonly found in dirhenium halide chemistry (Chapter 8). The primary and secondary molecules at a given crystallographic site are conformational enantiomers; although the populations of the two conformers at a given site are not equal, the crystals as a whole are racemic. However, with the use of a chiral phosphine ligand such as S,S-dppb [i.e., S,S-2,3-bis (diphenylphosphino)butane] chiral molecules can be obtained. The complexes `-Mo2X4(S,S-dppb)2 have been characterized by X-ray crystallography although several other well authenticated chiral molecules, such as `-Mo2Cl4(R-dppp)2 (R-dppp = R-1,2-bis(diphenylphosphino)propane), have not. A general discussion of the chiral character of these and other closely related dimolybdenum(II) complexes383,384,419,447-449 is given in Section 16.4.5.
Another interesting structural feature is seen in the case of _- and `-isomers of Mo2Cl4(dpdt)2 (dpdt = Ph2PCH2CH2P(p-tol)2), where because of the unsymmetric nature of the ligand these two isomers can exist in syn and anti forms. The anti-_-isomer has been crystallographically characterized (Table 4.9) and 1H NMR spectroscopy has been used to study the _- and `- forms.375,381 In the case of the _-isomers, a combination of the structural data and 1H NMR spectroscopy has been used382 to obtain the diamagnetic anisotropies of Mo–Mo quadruple bonds.
The large body of structural data now available on the _- and `-Mo2X4(LL)2 compounds clearly shows370,372,374,379 that there is an inverse linear relationship between the Mo–Mo bond distances and cos(2ρ), where ρ is the average torsional (twist) angle. This correlation is a direct consequence of the strength of the β component of the quadruple bond being a function of cos(2ρ).420 As ρ increases so the β component weakens. From this it follows that since the β Α β* transition energies are a function of β-bond strength, there should also be a relationship between the β Α β* electronic transition and cos(2ρ). This has been shown373,379 to be the case, and its further interpretation is discussed in Section 16.4.1.
The relative stabilities and interconversion of _ and ` isomers in solution have been studied. A unimolecular mechanism involving internal rotation of the Mo2 unit within the ligand cage is supported.377,417 Although the equilibrium constant may strongly favor the `-isomer in the _ ` equilibrium, the _-isomer can be obtained from the `-isomer by the use of a solvent system that permits selective precipitation of the _-form. This has been demonstrated through the conversion of `-Mo2Cl4(dpdt)2 to _-Mo2Cl4(dpdt)2.375 The case of _-Mo2X4(dppbe)2 (X = Cl or Br) is unusual in that the `-isomers have not been detected.385 The apparent failure to form `-Mo2X4(dppbe)2 is most likely a consequence of the rigidity of the dppbe ligand and its inability to bridge the two molybdenum atoms.
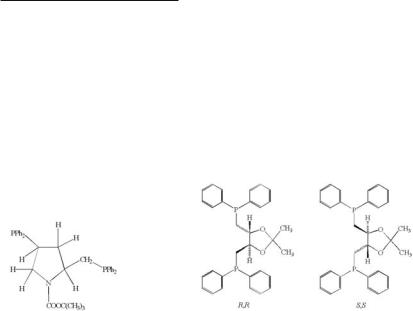
In addition to the complexes that contain bridging phosphine (and/or arsine) ligands and fiveor six-membered rings, a few examples are known of `-Mo2X4(LL)2 type compounds where the ring size is larger. The complex `-Mo2Cl4(dppp)2 (dppp or 1,3-dppp = Ph2P(CH2)3PPh2)376 contains two fused seven-membered rings, and has a disorder of the type where there are two perpendicular orientations of the Mo2 unit.387 The average twist angles for the primary and secondary orientations of the two independent molecules in the unit cell are close to 70°, reflecting this increase in ring size. There are two examples of structurally characterized dimolybdenum(II) complexes in which eight-membered rings are present. These are `-Mo2Cl4[(R,R)-diop]2 (the related isomer `-Mo2Cl4[(S,S)-diop]2 has also been prepared although its crystal structure has not been determined),386 and `-Mo2Cl4(S,S-bppm)2,389 both of which contain chiral phosphine ligands. Schematic representations of the diop ligand (as its R,R and S,S enantiomorphs) and
126Multiple Bonds Between Metal Atoms Chapter 4
S,S-bppm are given in 4.26 and 4.27. The conformational preference of each of these chiral ligands essentially effects an asymmetric synthesis and thereby produces only one of the possible configurational isomers. In these two instances, quite different twists are encountered, `-Mo2Cl4[(R,R)-diop]2 having a very large torsional angle (78°), while `-Mo2Cl4(S,S-bppm)2 is essentially eclipsed with each of the S,S-bppm ligands being bound through a phosphorus atom and its keto oxygen atom. The polydentate phosphine Ph2PCH2CH2P(Ph)CH2CH2P- (Ph)CH2CH2PPh2(tetraphos-1) reacts with K4Mo2Cl4 in methanol to give Mo2Cl4(tetraphos-1) in which the ligand has both bridging and chelating functionalities.426 The crystal contains the racemic R,R and S,S enantiomers.
4.26 |
4.27 |
The 31P{1H} NMR spectra of several of these complexes have been measured and generally consist of a singlet at room temperature, viz. Mo2Cl4(dmpm)2,367 Mo2X4(dppm)2 (X = Cl, Br or I),371,438 and _- and `-Mo2Cl4(dppee)2.380 In the case of _- and `-Mo2Cl4(dppee)2, the resonances are at β +35.9 and β +16.8 (spectra recorded in CD2Cl2),380 the upfield shift of the latter compound being typical of the greater shielding associated with six-membered rings compared to that of their five-membered analogs. The spectrum of `-Mo2I4(dppe)2 shows no signal at room temperature, but a broad resonance appears as the temperature is lowered and by −80 °C it is a sharp singlet.379 This temperature dependence is indicative of a low-energy fluxional process. For `-Mo2Cl4(S,S-bppm)2, singlets at β +32.5 and β −7.8 are assignable to the coordinated and free phosphine donor sites on the bppm ligand.389
The reaction chemistry of the Mo2X4L4 and Mo2X4(LL)2, compounds falls into two main categories, namely, non-redox ligand substitution reactions and redox chemistry in which the molybdenum unit is preserved. Ligand substitution reactions of the type Mo2X4L4 + 4L' Α Mo2X4L'4 + 4L have already been mentioned in the context of the synthetic strategies used to prepare tetrahalodimolybdenum(II) complexes. Halide substitution reactions have also been reported; the reaction of Mo2Cl4(dppm)2 with NaX (X = Br or I) in acetone has been used to prepare Mo2X4(dppm)2.438 In the reactions between Mo2X4(PBun3)4, where X = Cl or Br, and carboxylic acids, it was found107 that when Mo2X4(PBun3)4 and benzoic acid were reacted in refluxing benzene one of three complexes, viz. Mo2(O2CPh)2X2(PBun3)2, Mo2(O2CPh)4(PBun3)2 or Mo2(O2CPh)4, could be isolated depending upon the reaction conditions. Under similar conditions, alkyl carboxylic acids form only Mo2(O2CR)4.107 The crystal structure of Mo2(O2CPh)2Br2(PBun3)2 shows165 it to be centrosymmetric with a transoid arrangement of bridging benzoate ligands. The formation of Mo2(O2CPh)2Br2(PBun3)2 is similar to the reaction course that is encountered upon refluxing a mixture of 7-azaindole and Mo2Cl4(PEt3)4 in benzene.450 The emerald green complex Mo2(C7H5N2)2Cl2(PEt3)2 contains two monanionic 7-azaindolyl ligands and has a structure analogous to that of Mo2(O2CPh)2Br2(PBun3)2 although the Mo–Mo bond is distinctly longer (by c. 0.03 Å).

Molybdenum Compounds 127
Cotton
In a similar manner, the reactions between Mo2X4(PR3)4 (X = Cl or Br; PR3 = PEt3, PMe2Ph or PMePh2) and 2-hydroxy-6-methylpyridine (Hmhp) or 2,4-dimethyl-6-hydroxy- pyrimidine (Hdmhp) in toluene give Mo2(mhp)2X2(PR3)2 or Mo2(dmhp)2X2(PEt3)2.248 In the case of the mhp complexes, an alternative synthetic procedure is to react Mo2X4(PR3)4 with Mo2(mhp)4, and similar strategies can be used to prepare the complexes Mo2Cl3(mhp)(PR3)3 and Mo2Cl(mhp)3(PR3).248 An X-ray crystal structure determination has been carried out on cis-Mo2(mhp)2Cl2(PEt3)2.221
There are several reactions in which the Mo–Mo bond of Mo2X4L4 and Mo2X4(LL)2 is cleaved by /-acceptor ligands. While the reactions of Mo2X4(dppm)2 (X = Cl, Br or I) with an equivalent of RNC (R = Pri or But) in the presence of TlPF6 (in THF) or KPF6 (in acetone) give [Mo2X3(dppm)2(CNR)]PF6,438 an excess of RNC leads to seven-coordinate mononuclear complexes. The properties of [Mo2X3(dppm)2(CNR)]PF6 are in accord438 with a structure similar to that of the parent tetrahalo species.
A variety of electrochemical studies have demonstrated the relative ease with which phosphine containing complexes of the types Mo2X4L4 and Mo2X4(LL)2 undergo one-electron oxidations, which in some instances are reversible. Cyclic voltammetric measurements have been carried out on many of these complexes and the important results are summarized in Table 4.10. The first such study was carried out on solutions of Mo2Cl4(PR3)4 (R = Et or Prn), _-Mo2Cl4(dppe)2, and `-Mo2Br4(dppe)2 in 0.2 M (Bu4N)PF6–CH2Cl2 and revealed the presence of a quasi-revers- ible one-electron oxidation in the range +0.35 to +0.54 V versus SCE.451 Subsequently, a much more extensive range of complexes has been studied,248,367,379,380,385,438,439,452-456 with CH2Cl2 and THF used as solvents. No redox activity was observed for solutions of Mo2Cl4[P(OMe3)3]4 and Mo2Cl4[P(OMe)Ph2]4 in CH2Cl2.454 Since the E1/2(ox) values for solutions of Mo2X4(PR3)4 in THF are generally shifted by c. +0.3 V relative to those observed in CH2Cl2, this has permitted the observation of a one electron reduction (E1/2(red)) for several of these complexes when the former solvent is used. Occasionally, this process has been observed even in CH2Cl2 (Table 4.10) and, in the case of the complexes with bidentate phosphines, a one-electron reduction is readily accessible in both solvents. In some instances, measurements on the same complex have been carried out in independent studies in different laboratories. Generally, very similar results have been obtained. The data reported in Table 4.10 for Mo2Cl4(PMe2Ph)4 refer to the stable blue form (see above). A THF solution of the green form is said439 to have E1/2(ox) = +0.28 V and E1/2(red) = −0.95 V versus SCE under similar experimental conditions. The difference in potentials for these two forms seems too great in view of the close similarities of their other properties.
For the set of complexes Mo2X4(PMe3)4 (X = Cl, Br or I), the ease of oxidation and difficulty of reduction are both in the order Cl > Br > I. This ‘inverse’ halide order is opposite to that expected on electronegativity grounds, but it does reflect the tendency for low-valent iododes to be more stable than bromides, and these in turn more stable than chlorides.457 This order, which has been attributed to the effects of metal(d)-to-halide(d) back bonding,452 is also seen in the first oxidation (E1/2(ox) (1)) of the dirhenium(II) complexes Re2X4(PR3)4. The order Cl > Br for E1/2(ox) is followed for the other pairs of chloride/bromide complexes with monodentate phosphines, but this order is not so clear-cut in the case of the complexes that contain bidentate phosphines (see Table 4.10). For the pairs of _- and `-isomers of the type Mo2X4(LL)2, differences in the E1/2(ox) (or Ep,a) and E1/2(red) values380 do not provide a ready means of distinguishing between such isomers. For a series of chloride complexes, Mo2Cl4(PR3)4, a fairly good correlation was found454 to exist between the E1/2(ox) (or Ep,a) values and the β Α β* transition energies. These compounds become more difficult to oxidize as the electron withdrawing nature of the PR3 substituents increases and the β Α β* energy decreases.

128Multiple Bonds Between Metal Atoms Chapter 4
Table 4.10. Cyclic voltammetric data for dimolybdenum(II) complexes of the types Mo2X4L4 and Mo2X4(LL)2, X = Cl, Br, I, NCO or NCS)
Compound |
E1/2(ox) E1/2(red) |
Other |
Solvent |
Reference |
ref. |
|
processes |
electrode |
|||||
|
|
|
|
Mo2Cl4(PMe3)4
Mo2Br4(PMe3)4
Mo2I4(PMe3)4
Mo2Cl4(PEt3)4
Mo2Br4(PEt3)4
Mo2Cl4(PPrn3)4
Mo2Cl4(PBun3)4
Mo2Cl4(PH2Ph)4
Mo2Cl4(PMe2Ph)4
Mo2Br4(PMe2Ph)4
Mo2Cl4(PEt2Ph)4
Mo2Cl4(PHPh2)4
Mo2Cl4(PMePh2)4
Mo2Br4(PMePh2)4
Mo2Cl4(PEtPh2)4
Mo2Cl4(dmpm)2
Mo2Cl4(dppm)2
Mo2Br4(dppm)2
Mo2I4(dppm)2
_-Mo2Cl4(dppe)2
`-Mo2Cl4(dppe)2
_-Mo2Br4(dppe)2
`-Mo2Br4(dppe)2
`-Mo2I4(dppe)2
_-Mo2Cl4(dppee)2
`-Mo2Cl4(dppee)2
_-Mo2Br4(dppee)2
`-Mo2Br4(dppee)2
+0.74 |
−1.70 |
|
THF |
+0.77 |
−1.62 |
|
THF |
+0.47 |
|
|
CH2Cl2 |
+0.50 |
−1.72 |
|
CH2Cl2 |
+0.87 |
−1.48 |
|
THF |
+0.59 |
|
|
CH2Cl2 |
+0.88 |
−1.28 |
|
THF |
+0.96 |
−1.17 |
|
THF |
+0.73 |
−1.35 |
|
CH2Cl2 |
+0.67 |
−1.81 |
|
THF |
+0.35 |
|
|
CH2Cl2 |
+0.40 |
|
Ep,a = +1.43 |
CH2Cl2 |
+0.76 |
−1.59 |
|
THF |
+0.54 |
|
Ep,a = +1.44 |
CH2Cl2 |
+0.65 |
−1.89 |
|
THF |
+0.38 |
|
|
CH2Cl2 |
+0.64 |
−1.92 |
|
THF |
+0.38 |
|
|
CH2Cl2 |
5+1.2a,b |
|
|
CH2Cl2 |
+0.80 |
−1.63 |
|
THF |
+0.56 |
|
Ep,a = +1.50 |
CH2Cl2 |
+0.74 |
|
Ep,a = +1.47 |
CH2Cl2 |
+0.60 |
|
|
CH2Cl2 |
+0.92b |
|
|
CH2Cl2 |
+0.88b |
−1.54 |
|
THF |
+0.62 |
|
Ep,a = +1.69 |
CH2Cl2 |
+0.66 |
|
Ep,a = +1.5 |
CH2Cl2 |
+0.63b |
|
|
CH2Cl2 |
+0.49 |
−1.75c |
Ep,a = +1.25 |
CH2Cl2 |
+0.66 |
−1.5c |
|
CH2Cl2 |
+0.71 |
−1.28c |
|
CH2Cl2 |
+0.77b |
−1.03c |
Ep,a = +1.21 |
CH2Cl2 |
+0.61b |
−1.26 |
|
CH2Cl2 |
+0.59 |
−1.37 |
|
CH2Cl2 |
+0.65b |
−1.15 |
|
CH2Cl2 |
+0.59 |
−1.07 |
|
CH2Cl2 |
+0.62 |
−1.04c |
|
CH2Cl2 |
+0.58b |
−1.18 |
|
CH2Cl2 |
+0.75b |
−1.29 |
|
CH2Cl2 |
+0.64b |
−1.04 |
|
CH2Cl2 |
+0.77b |
−1.07 |
|
CH2Cl2 |
SCE |
452d |
Ag/AgCl |
399 |
SCE |
452e |
Ag/AgCl |
399 |
SCE |
452 |
SCE |
452 |
SCE |
452 |
Ag/AgCl |
399 |
Ag/AgCl |
399 |
SCE |
452 |
SCE |
451,452e |
Ag/AgCl |
248 |
SCE |
452 |
Ag/AgCl |
248 |
SCE |
452 |
SCE |
451,452e |
SCE |
433 |
SCE |
433c,f |
Ag/AgCl |
454 |
SCE |
439 |
Ag/AgCl |
248e |
Ag/AgCl |
248 |
Ag/AgCl |
454 |
Ag/AgCl |
454 |
SCE |
439 |
Ag/AgCl |
248e |
Ag/AgCl |
248 |
Ag/AgCl |
454 |
Ag/AgCl |
367 |
Ag/AgCl |
438 |
Ag/AgCl |
438 |
Ag/AgCl |
438 |
Ag/AgCl |
380g |
Ag/AgCl |
380 |
Ag/AgCl |
380 |
Ag/AgCl |
380g |
Ag/AgCl |
379 |
Ag/AgCl |
380 |
Ag/AgCl |
380 |
Ag/AgCl |
380 |
Ag/AgCl |
380 |