

Organicheskaya_khimia
.pdfПостулат Дж. Хэммонда
Для косвенной оценки строения переходного состояния используют
постулат Дж. Хэммонда (1955): несущественные энергетические изменения
сопровождаются незначительными изменениями молекулярной структуры. Более понятно можно сформулировать так: строение переходного состояния
похоже на строение тех веществ, к которым оно (переходное состояние) ближе по энергии. В экзотермических реакциях переходное состояние ближе по строению к исходным реагентам (рис. 10). Такой активированный комплекс называют ранним переходным состоянием. Переходное состояние в эндотермических реакциях ближе по структуре к продуктам реакции, его называют поздним. Однотипные воздействия на похожие структуры приводят к близкому результату. Поэтому все факторы, стабилизирующие (понижающие энергию) энергетически близкое к переходному состоянию (исходное, промежуточное или конечное) вещество, понижают и энергию
активированного комплекса.
Использование постулата Хэммонда особенно полезно при рассмотрении многостадийных реакций (рис. 11).
E
[ПC1]
[ПC2]
Ea1 |
Ea2 |
|
Промежуточный
продукт
(интермедиат)
Исходные |
Продукты |
|
Координата реакции
Рис 11. Энергетическая диаграмма двухстадийной реакции
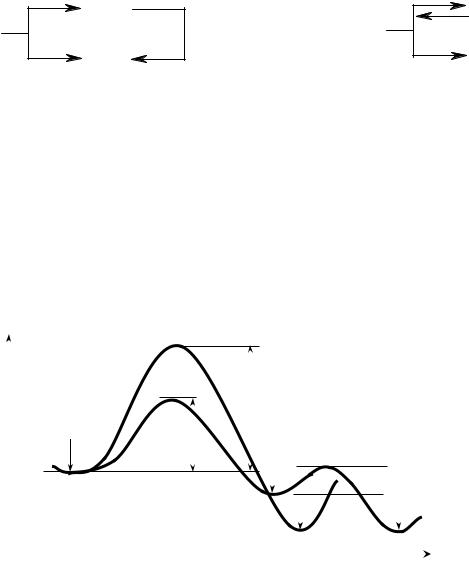
Из рис. 11 видно, что реакция протекает в две стадии, через один промежуточный продукт. Превращение продуктов в интермедиат (первая стадия) имеет большее значение для всей реакции, чем превращение интермедиата в продукты реакции (вторая стадия). В этом убеждают соответствующие энергии активации первой и второй стадий (Ea1 и Ea2 соответственно). Весь ход реакции определяет её высшая энергетическая точка
– переходное состояние первой стадии [ПС1]. Если применить к этой реакции постулат Хэммонда, легко сделать вывод, что к переходным состояниям обеих стадий реакции энергетически ближе всего промежуточный продукт.
21

Важно отметить: во многих случаях интермедиаты могут быть зарегистрированы физическими методами и, следовательно, их строение может быть известно. Строение же переходного состояния неизвестно никогда.
Зная строение интермедиата, мы в дальнейшем сможем делать заключение о том, какие структурные элементы могут его стабилизировать, а какие дестабилизировать, и переносить это заключение на энергию переходного состояния.
•Равновесные реакции, константа равновесия, кинетический и термодинамический контроль. Механизм реакции и пути его установления. Принцип микроскопической обратимости, постулат Хэммонда.
•Кислоты и основания в органической химии (теории Бренстеда-Лоури и Льюиса). Сопряженные кислоты и основания. Кислотно-основные равновесия. Константа кислотности (рКа), константа основности (рКb). Влияние заместителей на кислотность и основность органических соединений.
1.10. Равновесные реакции
Особенность многих органических реакций состоит в том, что они равновесные.
aA + bB = dD + fF
Скорость прямой реакции: Vпр = KпрCAaCBb Скорость обратной реакции: Vоб = KобCDdCFf
Когда скорости прямой и обратной реакции равны, наступает равновесие:
Vпр = Vоб
KпрCAaCBb = KобCDdCFf
Kпр |
= Kравн = |
[D]d[F]f |
|
|
[A]a[B]b |
||
Kобр |
|||
|
Кроме того, константу равновесия можно выразить через свободную энергию:
G = -RTlnKравн
Важно: при наступлении состояния равновесия прямая и обратная реакции не прекращаются. Одновременно протекают прямая и обратная реакция, но из-за постоянства концентраций исходных веществ и продуктов, кажется, что все процессы остановились. Просто скорости прямой и обратной реакции равны!
22
Если Vпр > Vобр, то в равновесном состоянии концентрации продуктов будут выше концентраций исходных веществ. Равновесие сдвинуто вправо. При Vпр < Vобр равновесие смещено влево, в сторону исходных веществ. Если в первом случае не удается выделить исходные соединения, считают, что реакция полностью прошла. Во втором случае - если не удается обнаружить продукты - реакция не идет.
1.11.Кинетический и термодинамический контроль
Вряде случаев в результате протекания параллельных реакций в реакционной массе могут образовываться два продукта (C и D), один из которых превращается в другой либо непосредственно (по реакции 3) (рис 12а), либо в результате обратимости первой реакции (рис. 12б). Какой из продуктов является основным, очень часто зависит от температурных условий проведения превращения.
|
1 |
|
|
1 |
|
C |
|
C |
|
|
|
|
||
A + B |
|
3 |
A + B |
3 |
2 |
|
|||
|
D |
|
D |
|
|
|
|
||
|
|
|
|
2 |
|
a) |
|
|
б) |
Рис. 12. Схемы конкурентных реакций Энергетическая диаграмма процессов, изображенных на рис. 12а,
показывает, что образование продукта С происходит быстрее, т.к. ЕаС этой реакции заметно меньше, чем ЕаD (рис. 13). При низкой температуре, когда энергия реагирующих молекул невелика, основное направление реакции – образование соединения С. Этот процесс контролируется скоростью и называется кинетически контролируемой реакцией, а соединение С –
продуктом кинетического контроля.
EaD
Е
|
C |
|
A + B |
D |
D |
|
EaC |
|
|
|
EaC-D |
координата реакции
Рис. 13. Кинетический и термодинамический контроль (последовательное превращение)
23
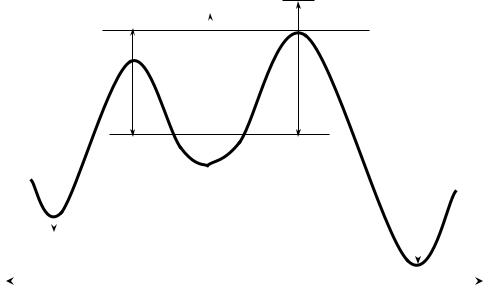
Образование соединения D требует большей энергии, поэтому при низкой температуре оно – побочный продукт. Однако продукт D термодинамически более устойчив, чем С. Поэтому с ростом температуры проведения реакции, когда большее число молекул способно перебраться через высокий энергетический барьер, в реакционной массе наблюдается преобладание продукта D. Процесс образования соединения D контролируется энергетическими факторами и называется термодинамически контролируемой реакцией. Соединение D – продукт термодинамического контроля.
Соединение С образуется быстро. Его стабильность относительно низка, а энергия активации превращения С в D (ЕаС-D) невелика. При проведении реакции при повышенной температуре (т.е. в условиях термодинамического контроля) происходит превращение соединения С в более энергетически выгодный продукт Д.
На рис. 14 показаны энергетические профили реакций кинетического (образование С) и термодинамического (образование D) контроля для обратимых превращений.. Причины особенностей протекания реакций аналогичны изложенным выше.
Е
C |
EaD |
EaD |
|
D |
|
|
|
|
|
|
A + B |
Ход реакции |
Ход реакции |
кинетического контроля |
термодинамического контроля |
Рис. 14. Кинетический и термодинамический контроль (обратимое превращение)
Отличается только превращение продукта кинетического контроля С в продукт термодинамического контроля D. При повышенной температуре происходит превращение соединения С в исходные вещества A и B. Процесс протекает быстро, т.к. энергия активации этого процесса незначительна. Превращение D в исходные продукты протекает значительно труднее, и он накапливается в реакционной смеси.
24

Образование продуктов кинетического и термодинамического контроля – классический случай протекания органических реакций. Конкретные реакции будут рассмотрены в соответствующих разделах (см., например, химические свойства 1,3-алкадиенов).
1.12. Кислотно-основные свойства органических соединений
В органической химии используются две теории кислот и оснований:
•теория Брёнстеда-Лоури;
•теория Льюиса.
Кислоты и основания по Брёнстеду-Лоури
Воснове теории - перенос протона (Н+). Согласно Бренстеду-Лоури кислоты – вещества, способные выступать в качестве донора протона (т.е. его отдавать); основания – вещества, способные быть акцепторами протона (т.е. его присоединять).
Уравнение кислотно-основного взаимодействия по Брёнстеду-Лоури:
A-H |
+ |
:B |
|
|
|
A- |
+ |
B-H+ |
|
|
|||||||
|
|
|
||||||
кислота |
|
основание |
|
|
сопряженное |
|
сопряженная |
|
|
|
|
|
|
|
основание |
|
кислота |
Для проявления кислотных свойств органических соединений связь водород-элемент должна быть полярной или поляризованной. Многие вещества содержат связи О-Н, N-H, S-H. В ряде случаев кислотные свойства могут проявлять и С-Н фрагменты. В зависимости от элемента, отдающего протон, органические кислоты классифицируют на ОН-, SH-, NH- и СН-кислоты. Кислотные свойства могут проявлять нейтральные молекулы или катионы.
Основания же должны иметь или неподеленную пару электронов у атома, присоединяющего протон, или отрицательный заряд. В некоторых случаях основные свойства могут проявлять и соединения с π-связями.
Силу кислоты выражают количественно через константу кислотности:
[A-][H3O+]
Ka = Kp[H2O] =
[AH]
где Ka – константа кислотности; Kp – константа равновесия.
Кислота там сильнее, чем больше константа кислотности. Часто пользуются значениями рКа. Чем меньше величина рКа, тем сильнее кислота.
рКа = -lgКа
25

Например, рКа фенола = 10, рКа этанола = 16. Это означает, что фенол на шесть порядков (в миллион раз) более сильная кислота, чем этиловый спирт.
Основность вещества может быть выражена через рКb.
рКb = 14 - рКa
Важно помнить, что рКа воды = 15,7. Все вещества, которые имеют рКа больше, чем вода, не способны проявлять кислые свойства в водных растворах. Вода, как более сильная кислота, подавляет диссоциацию более слабых кислот. Так как у большинства органических соединений кислотные свойства выражены во много раз слабее, чем у воды, разработан полярографический подход к оценке их кислотности (И.П. Белецкая и др.). Он позволяет оценивать кислотность до рКа = 50, хотя для очень слабых кислот значения рКа можно оценить только очень приблизительные.
Чрезвычайно важна качественная оценка кислотности как в рядах близких по строению веществ, так и для соединений различных классов. Способность кислоты отдавать протон связана со стабильностью образующегося аниона. Чем стабильнее образующийся анион, тем меньше его стремление захватить обратно протон и превратиться в нейтральную молекулу. При оценке относительной стабильности аниона надо учитывать несколько факторов.
1) Природа атома, отдающего протон. Атом тем легче теряет протон, чем выше его электроотрицательность и поляризуемость. Поэтому в ряду кислот способность к диссоциации уменьшается следующим образом:
S-H > O-H > -`-H > C-H
Этот ряд прекрасно соответствует свойствам атомов, известным из периодической таблицы.
2) Влияние окружения. Если сравниваются близкие по строению вещества, оценка проводится сравнением электронной плотности на атоме, отдавшем протон. Все структурные факторы, способствующие уменьшению заряду, стабилизирует анион, а увеличению заряда – дестабилизируют. Таким образом, все акцепторы увеличивают кислотность, все доноры – уменьшают.
|
R - Z- |
|
|
R - Z- |
|
|
|
|
|
||
стабилизация |
дестабилизация |
||||
Это происходит независимо от того, за счет какого эффекта передачи электронов (индуктивного или мезомерного) происходит перераспределение электронной плотности.
3)Сольватационный эффект. Сольватация (взаимодействие с
молекулами растворителя) повышает стабильность аниона за счет
26
перераспределения избытка электронной плотности между анионом и молекулами растворителя. В общем случае закономерность следующая:
•чем полярнее растворитель, тем сильнее сольватация;
•чем меньше ион, тем лучше он сольватируется.
Основность по Брёнстеду – способность вещества предоставить свою пару электронов для взаимодействия с протоном. Как правило, это вещества, содержащие в молекуле атомы азота, кислорода и серы.
Чем слабее основный центр удерживает пару электронов, тем выше основность. В ряду
R3-` > R2O > R2S
основность уменьшается. Эту последовательность легко запомнить, используя мнемоническое правило “NOS”.
Среди оснований Брёнстеда существует зависимость: анионы более сильные основания, чем соответствующие нейтральные молекулы. Например, гидроксид-анион (–ОН) более сильное основание, чем вода (Н2О). При взаимодействии основания с протоном могут образовываться ониевые катионы:
•R3О+ - оксониевый катион;
•NR4+ - аммониевый катион;
•R3S+ - сульфониевый катион.
Качественная оценка основности у близких по строению веществ проводится с использованием той же логики, что и оценка кислотности, но с обратным знаком.
Поэтому все акцепторные заместители основность уменьшают, все донорные – увеличивают.
Кислоты и основания по Льюису
Основания по Льюису – доноры электронной пары, как и основания по Брёнстеду.
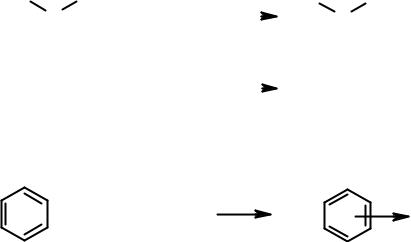
Определение Льюиса для кислот заметно отличается от привычного (по Брёнстеду). Кислотой по Льюису считается любая молекула или ион, имеющая свободную орбиталь, которая может быть в результате взаимодействия заполнена электронной парой. Если по Брёнстеду кислота – донор протона, то по Льюису сам протон (Н+) – кислота, поскольку его орбиталь пуста. Кислот Льюиса очень много: Na+, Mg2+, SnCl4, SbCl5, AlCl3, BF3, FeBr3 и т.д. Теория Льюиса позволяет описать многие реакции как кислотно-основные взаимодействия. Например:
27
|
N : |
|
|
+ |
|
- |
|||||||
|
+ |
BF3 |
|
|
|
N |
|
|
|
BF3 |
|||
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
||||
H3C |
|
Cl |
|
AlCl3 |
+ |
- |
|||||||
|
+ |
|
|
H3C |
|
Cl |
|
AlCl3 |
|||||
|
|
|
|
|
|||||||||
Часто в реакциях с кислотами Льюиса в качестве оснований участвуют органические соединения, являющиеся донорами пары π-электронов:
+ +NO2 |
+ |
NO2 |
В органической химии принято следующее:
•если используется термин «кислота» - подразумевается кислота по Брёнстеду;
•если используют термин «кислота» в льюисовском понимании – говорят «кислота Льюиса».
28
2.Алифатические углеводороды
•Алканы. Гомологический ряд, номенклатура, изомерия, алкильные радикалы. Электронное строение молекул алканов, sp3-гибридизация, σ-
связь. Длины C-C и C-H связей, валентные углы, энергии связей.
Пространственная изомерия органических веществ. Способы изображения пространственного строения молекул с sp3- гибридизованными атомами углерода. Спектральные характеристики алканов. Физические свойства алканов и закономерности их изменения в гомологическом ряду.
2.1.Алканы (насыщенные ациклические соединения,
парафины)
Алканы - углеводороды с открытой цепью атомов, отвечающие формуле СnH2n+2, где атомы углерода связаны между собой только σ-связями.
Термин «насыщенный» означает, что каждый углерод в молекуле такого вещества связан с максимально возможным числом (с четырьмя) атомов.
Строение метана подробно изложено выше.
Изомерия
CH4 |
H3C |
|
CH3 |
H C |
CH |
3 |
|
||||||
|
|
|
|
3 |
|
|
метан |
этан |
пропан |
|
|||
|
|
CH |
|
H3C |
|
CH3 |
|
|
|
|
|
H3C |
3 |
|
|
|
|
|
|
бутаны |
|
|
|
|
|
CH3 |
|
|||||
|
|
|
|
|
|
|
|
|||
|
|
|
|
CH3 |
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
H3C |
CH3 H3C |
|
|
H3C |
|
пентаны |
||||
|
|
|
|
|||||||
|
|
|
|
CH3 |
|
|
|
CH3 |
|
|
Три первых члена гомологического ряда (метан, этан и пропан) существуют в виде одного структурного изомера. Начиная с бутана число изомеров стремительно растет: у пентана три изомера, а у декана (С10Н22) их уже 75.
Конформационная изомерия
Стереоизомеры (пространственные изомеры) – вещества, имеющие одинаковый состав и последовательность томов, но различное расположение
29
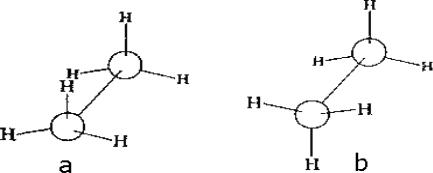
их в пространстве. σ-Связь симметрична относительно оси, связывающей центры атомов, и при повороте фрагментов относительно друг друга энергия связи не меняется. Энергетически неравноценные формы молекул, переходящие друг в друга только за счет свободного вращения или изгиба связей, называются конформациями.
Конформация а (рис. 15) называется заслоненной, конформация b – заторможенной. Возможно бесконечное количество промежуточных (скошенных) конформаций.
Рис. 15. Заслоненная (а) и заторможенная (b) конформации этана
Удобно изображать конформации с помощью проекций Ньюмена – взгляд направлен вдоль связи, относительно которой происходит вращение. В заслоненной конформации атомы находятся ближе друг к другу, чем в заторможенной. Возникает сила, стремящаяся повернуть один фрагмент относительно другого – торсионное напряжение. Заторможенная конформация поэтому энергетически более выгодна, чем заслоненная. В этане энергетический барьер между конформациями составляет всего 3 ккал/моль (12 кДж/моль) (рис. 16).
30