

ТОХФ / 1 группа (ХТУМ) / Инструментальные методы анализа - Гречишкина / хроматография
.doc
5. Хроматографические методы разделения и анализа
Хроматографические методы относятся к молекулярному уровню исследования веществ. Цель методов этого уровня – определить, какие именно молекулы и в каком количестве содержатся в изучаемом объекте.
Х орошие
результаты получаются при анализе
молекулярными методами жидких образцов,
поскольку в этом случае нет проблем с
приготовлением представительных проб.
На молекулярном уровне изучают нефти
и нефтепродукты, гуминовые вещества,
гидрогенизаты, экстракты из твердых
объектов, битумоиды, воски, смолы и
другие продукты переработки природных
энергоносителей (рис. 5.1).
орошие
результаты получаются при анализе
молекулярными методами жидких образцов,
поскольку в этом случае нет проблем с
приготовлением представительных проб.
На молекулярном уровне изучают нефти
и нефтепродукты, гуминовые вещества,
гидрогенизаты, экстракты из твердых
объектов, битумоиды, воски, смолы и
другие продукты переработки природных
энергоносителей (рис. 5.1).
Рис. 5.1. Хроматограмма газойля, полученная методом капиллярной газовой хроматографии
Твердые вещества затруднительно изучать на этом уровне, поскольку извлечение молекул зачастую нецелесообразно из-за сложности процесса и непредставительности проб. Как правило, такие объекты перед анализом приходится либо подвергать дополнительной обработке, например, фракционированию (в том числе деструктивному), а затем анализировать полученные фракции, либо анализировать более общие, чем структура конкретной молекулы, параметры, получаемые молекулярными методами (соотношение Салиф и Саром, определенное методом ЯМР 13С, и т.п.).
Природные энергоносители и продукты их переработки – смеси сложного состава. Например, нефть содержит порядка 300-350 различных веществ, поэтому прежде применения молекулярных методов исследования обычно проводят предварительную переработку с целью фракционирования даже жидких объектов. С этой целью используют экстракцию селективными растворителями, разнообразные разгонки, препаративную высокоэффективную жидкостную хроматографию (ВЭЖХ).
В результате этой обработки обычно получается фракция исходного объекта – смесь с меньшим количеством веществ, иногда удается выделить из смеси чистые вещества. В ряде случаев нет необходимости получать для анализа молекулярными методами химически чистые вещества. Аналитическая хроматография, например, служит одновременно методом разделения и анализа.
Хроматография – это метод разделения компонентов смеси, основанный на различии в равновесном распределении их между двумя несмешивающимися фазами, одна из которых неподвижна, а другая подвижна. Компоненты образца движутся по колонке, когда они находятся в подвижной фазе, и остаются на месте, когда находятся в неподвижной фазе. Чем больше сродство компонента к неподвижной фазе и чем меньше – к подвижной, тем медленнее он движется по колонке и тем дольше в ней удерживается. За счет различия в сродстве компонентов смеси к неподвижной и подвижной фазам достигается основная цель хроматографии – разделение смеси за приемлемый промежуток времени на отдельные полосы (пики) компонентов по мере их продвижения по колонке с подвижной фазой.
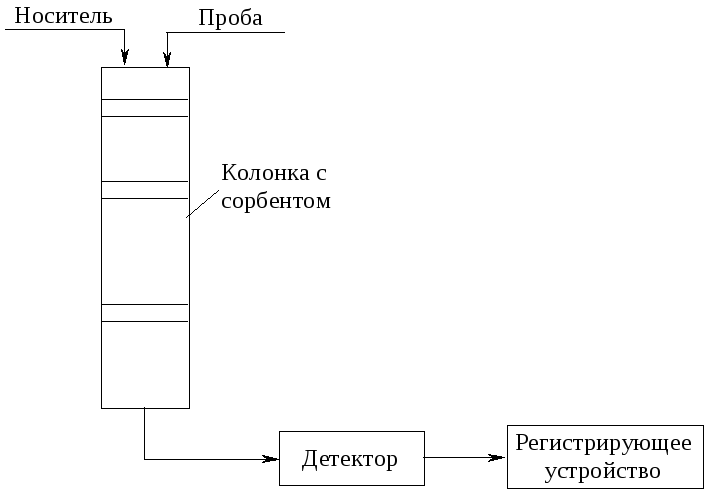
Блок-схема хроматографа представлена на рис. 5.2. Разделение компонентов смеси происходит в хроматографической колонке, заполненной сорбентом (неподвижная фаза). Проба вводится в жидком или газообразном виде в начало колонки и продвигается по ней с потоком подвижной фазы (носителя, элюента). На выходе из колонки поток состоит из зон, содержащих последовательно разделенные компоненты, перемежающиеся зонами, содержащими чистый носитель. Этот поток поступает в детектор, где определяется наличие и концентрация выделенного компонента. Результаты детектирования выводятся на регистрирующее устройство (компьютер, самописец).
Если при вводе пробы какие-то компоненты находятся в твердом виде, они будут отфильтрованы и не будут участвовать в хроматографическом процессе. Точно так же компоненты, не взаимодействующие с неподвижной фазой, пройдут через колонку с подвижной фазой, не разделяясь на компоненты.
|
|
|
Рис. 5.2. Блок-схема хроматографа |
В случае протекания хроматографического процесса без нарушений можно получить хроматограммы вида а, б или в (рис. 5.3), которые иллюстрируют хроматографические разделения, отличающиеся эффективностью (а и б) при равной селективности и селективностью (б и в) при равной эффективности. Эффективность колонки тем выше, чем у́же пик получается при том же времени удерживания. Селективность колонки растет при увеличении отношения времен удерживания компонентов.
Важным параметром разделения пиков является разрешение R, которое увеличивается по мере возрастания селективности, отражаемой ростом числителя, и роста эффективности, отражаемой снижением значения знаменателя из-за уменьшения ширины пиков. Величину R можно определить по хроматограмме (рис. 5.4):
![]() или
или
![]()
Использование той или иной формулы зависит от того, насколько возможно точно определить ширину пика у его основания. Довольно часто пики имеют размытый задний фронт, в этом случае лучше пользоваться вторым вариантом формулы. Пригодным для использования считается разделение с разрешением не менее 1, допустимо использование в количественных расчетах хроматограмм с R0,5. В противном случае сильно возрастает погрешность расчетов.
|
|
|
Рис. 5.3. Вид хроматограммы в зависимости от эффективности и селективности колонки: а – обычная селективность при пониженной эффективности; б – обычные селективность и эффективность; в – обычная эффективность при повышенной селективности
|
|
|
|
Рис. 5.4. Параметры удерживания хроматографического пика: tR – время удерживания пика; t0 – время удерживания несорбируемого компонента; h – высота пика; W1/2 – ширина пика на половине его высоты; W1, W2 – ширина хроматографических пиков |


Параметром удерживания, характеризующим природу анализируемого вещества, является коэффициент емкости:
![]() .
.
Коэффициент емкости позволяет сравнивать по эффективности и селективности различные колонки при прочих равных условиях анализа данного вещества.
Для количественного анализа требуется измерить площади или высоты пиков, а затем рассчитать количественный состав смеси на основании хроматографических данных. Высоту пиков можно использовать в качестве измеряемого параметра в случае очень узких пиков, так как при этом слишком велика погрешность определения ширины пика. В случае широких пиков измерения ведут по их площадям.
Количественная интерпретация хроматограмм может проводиться различными методами:
-метод абсолютной калибровки;
-метод внутреннего стандарта;
-метод внутренней нормализации и др.
Метод абсолютной калибровки или внешнего стандарта состоит в следующем: готовят несколько стандартных калибровочных растворов с различными известными концентрациями определяемых компонентов. В хроматографическую колонку вводят определенный объем каждого стандартного раствора и измеряют площадь или высоту пика. Для каждого вещества строят график зависимости высоты или площади пика от концентрации или массы определяемого вещества (т.е. калибровочную кривую). Затем в хроматограф вводят тот же объем исследуемой пробы. После измерения полученных пиков, пользуясь калибровочной кривой для определяемого компонента, находят концентрацию вещества, соответствующую полученной площади (высоте) пика.
Метод внутреннего стандарта основан на добавлении одинакового объема вещества (внутреннего стандарта) с известной концентрацией, не присутствующего в первоначальной смеси, в неизвестный образец и в калибровочные растворы для получения отдельного пика на хроматограмме. Это вводимое вещество играет роль внутреннего маркера и компенсирует влияние небольших отклонений параметров разделения на размер пиков.
В этом методе калибровки измеряется отношение площади (высоты) определяемого пика и пика внутреннего стандарта и строится график зависимости этого отношения от концентрации вещества в калибровочных растворах. Этим графиком пользуются для определения концентрации определяемого компонента в исследуемой пробе аналогично методу абсолютной калибровки.
Внутренний стандарт должен быть химически инертным, полностью отделяться от других компонентов смеси, концентрация его должна быть близка к концентрации определяемых веществ. По возможности внутренний стандарт должен быть аналогичен по структуре определяемым веществам.
Метод нормировки (внутренней нормализации) основан на измерении площади (высоты) каждого пика в хроматограмме и вычислении процентного содержания каждого компонента, пропорционального суммарной площади (сумме высот). Содержание всех компонентов принимают равным 100%. Такой подход используют после определения поправочных коэффициентов на отклик детектора для каждого вещества и после умножения площади (высоты) пика на соответствующий коэффициент, чтобы учитывать различную чувствительность детектора к каждому компоненту смеси.
Существует большое количество вариантов хроматографического анализа, причем классифицировать их можно различными способами.
По масштабу хроматографию можно разделить на препаративную, аналитическую, микроколоночную и капиллярную. В этом ряду уменьшается диаметр колонки (с 20-25 мм для препаративной до 0,2 мм для капиллярной), размер частиц сорбента (наполнителя колонки), объем вводимой пробы (с граммов или миллилитров в препаративной хроматографии до десятых долей микролитра в капиллярной), но эффективность и селективность разделения в этом ряду увеличиваются. Препаративная хроматография предназначена для выделения химически чистых веществ из смеси в количестве, достаточном для дальнейшего использования либо в синтетических целях, либо для анализа другими методами. Остальные разновидности хроматографии предназначены только для анализа смесей. Колонки обычно представляют собой металлическую, стеклянную или кварцевую трубку длиной от нескольких сантиметров (микроколоночная хроматогафия) до нескольких дециметров, заполненную сорбентом. Особняком стоит капиллярная хроматография, в которой колонки представляют собой длинный (20-50 м длиной) полый капилляр, на стенки которого нанесена неподвижная фаза.
По носителю хроматография бывает газожидкостная (часто называется просто газовой) и жидкостная. Разновидностью хроматографии можно считать капиллярный электрофорез.
В газовой хроматографии носителем является газ, подаваемый под определенным давлением из баллона или другого источника (генератор водорода, компрессор). Вещество пробы взаимодействует в основном с жидкой фазой, нанесенной на сорбент. Природой этой фазы определяется разделяющая способность колонки. Кроме того, на разделение компонентов смеси можно влиять, задавая определенный температурный режим анализа. Поскольку детектирование веществ ведется в газовой фазе, от режима испарения пробы зависит результат анализа.
В жидкостной хроматографии носителем (элюентом) является жидкость, подаваемая насосом под определенным давлением (вариант ВЭЖХ, жидкостная хроматография без давления не используется в инструментальном анализе). Существует несколько разновидностей жидкостной хроматографии: нормально-фазная, обращенно-фазная, ионобменная, гель-хроматография и др. Наиболее широко применяются первые два варианта.
В нормально-фазной хроматографии элюентом служит неполярное вещество или смесь веществ с невысокой полярностью (гексан, дихлорэтан, серный эфир и т.п.), а сорбентом – полярное вещество, например, силикагель. В обращенно-фазной хроматографии используют полярный элюент (вода, метанол, ацетонитрил и т.п.), а сорбентом обычно служит силикагель с химически привитыми неполярными (как правило, длинными алкильными или алкилсилильными) группами. В жидкостной хроматографии вещество пробы взаимодействует как с сорбентом колонки, так и с элюентом, этим определяется порядок выхода компонентов смеси из хроматографической колонки. В нормально-фазной хроматографии полярные вещества будут сильнее взаимодействовать с полярным сорбентом, и в результате они дольше удержатся в колонке, т.е. вещества будут выходить в порядке увеличения полярности. В обращенно-фазной хроматографии из-за обратного соотношения полярностей сорбента и элюента будет наблюдаться обратный порядок выхода компонентов смеси из колонки: первыми выходят самые полярные соединения, а затем остальные в порядке уменьшения полярности.
При анализе методом ВЭЖХ прежде всего требуется выбрать, в каком именно варианте будет проводиться анализ: на нормальной или обращенной фазе, затем выбирают химическую природу сорбента. На эффективность разделения в жидкофазной хроматографии сильно влияет состав элюента, который обычно представляет собой смесь двух или более растворителей, причем имеет значение, как химическая природа растворителя, так и количественное соотношение компонентов элюента.
Электрофорез как инструментальный метод анализа существует только в капиллярном варианте. Капилляр представляет собой длинную (20-50 м) тонкую (диаметр 0,2-0,5 мм) полую трубку, на стенки которой изнутри нанесена неподвижная фаза (сорбент). Капилляр заполнен буферным раствором, возможно введение различных добавок, повышающих эффективность процесса разделения. Движущей силой этого процесса является разность потенциалов порядка 30 кВ, приложенная к концам капиллярной колонки. Вещество пробы взаимодействует с сорбентом, с буфером, с добавками. Изменяя состав, концентрацию и pH буфера, природу сорбента, химический состав и концентрацию добавок, можно влиять на разделение компонентов смеси.
Различные молекулы несут на себе различные заряды и поэтому при наложении электрического поля могут ускоряться в различной степени, поэтому они перемещаются в растворе буфера с различной скоростью. Незаряженные молекулы разделяют с помощью мицеллярного капиллярного электрофореза. В этом случае к буферу добавляется поверхностно активное вещество. Нейтральные молекулы распределяются между буфером и мицеллами в соответствии с их гидрофобностью. В то время как электрофорез обусловливает разделение частиц с различной подвижностью, течение буферного раствора в электрическом поле определяется электроосмосом. Это явление позволяет проводить разделение катионных и анионных соединений в одном анализе, так как одни ионы двигаются быстрее осмотического потока, ионы противоположного заряда будут двигаться медленнее, незаряженные частицы будут передвигаться вместе с осмотическим потоком.
Разные методы хроматографии применимы для различных типов веществ. Например, возможности газовой хроматографии позволяют анализировать вещества с молекулярной массой не более 250-300 у.е. Это ограничение возникает из-за того, что детектирование соединений ведется в газовой фазе, т.е. компоненты смеси требуется испарить, для чего приходится нагревать колонку. Современные фазы для газовой хроматографии выдерживают температуры не выше 400С, поэтому возможен анализ только тех веществ, температура испарения которых не превышает 300-350С. Кроме того, методом газовой хроматографии нельзя анализировать те соединения, которые разлагаются при нагревании.
Возможности жидкостной хроматографии ограничиваются, в основном, типом применяемого сорбента и детектора. Этим методом можно анализировать вещества с молекулярной массой до 1000 у.е. и даже несколько выше. Меняя условия анализа в рамках этого метода всегда можно найти условия, в которых данную смесь компонентов удастся разделить достаточно хорошо.
Метод капиллярного электрофореза подходит только для молекул, имеющих возможность перераспределения заряда внутри молекулы и образующих диполь в электрическом поле. С учетом различных приемов, (например добавок в буферный раствор), этим методом не удается разделить только алканы нормального строения, для остальных веществ в широком диапазоне молекулярных масс удается подобрать условия эффективного анализа.
В хроматографии может использоваться большое количество разнообразных детекторов в зависимости от метода и целей анализа.
В газовой хроматографии широко применяется катарометр – детектор общего назначения, проводящий определение концентраций веществ по их теплопроводности. Для анализа органических соединений чаще всего используется пламенно-ионизационный детектор (ПИД), в котором газ, выходящий из колонки, смешивается с водородом, сгорает и ионизируется, а детектирование компонентов смеси ведется по электропроводности пламени. Электроно-захватный детектор, в котором источником ионизации в отличие от ПИД служит тритий, предназначен для галогенсодержащих соединений, по которым он обладает максимальной чувствительностью. По этой же причине пламенно-фотометрический детектор, в котором определение ведется по фотометрии пламени, используется для серосодержащих соединений.
Для жидкостной хроматографии и капиллярного электрофореза используются одни и те же детекторы. Принципы их действия основаны на методах анализа молекулярного уровня, например, на спектроскопии в инфракрасной (ИК), видимой и ультрафиолетовой (УФ) области, масс-спектроскопии и др.
Наиболее широко распространены УФ-детекторы с переменной длиной волны, работающие на одной выбранной волне или проводящие сканирование по всему диапазону, и диодно-матричные, позволяющие в каждый момент времени снимать весь спектр значений во всем диапазоне длин волн. Эти детекторы пригодны для анализа веществ, поглощающих излучение в УФ-области. Рефрактометры (детекторы, проводящие определение веществ по их показателю преломления) и масс-селективные детекторы (масс-спектрометры) являются приборами общего назначения, т.е. подходят для любых соединений. Только вещества, обладающие флуоресценцией, можно анализировать при помощи флуориметра. Детектором общего назначения является также ИК-Фурье детектор, но при помощи этого детектора можно выбрать класс только тех соединений, который будет регистрироваться, в то время как остальные соединения не будут давать сигнала.