

ТОХФ / 1 группа (ХТУМ) / Инструментальные методы анализа - Гречишкина / Опт. спектр
.1.doc
![]() 6.
Молекулярная спектроскопия в УФ-, видимой
и ИК-области
6.
Молекулярная спектроскопия в УФ-, видимой
и ИК-области
Оптические спектральные методы молекулярного уровня основаны на взаимодействии электромагнитного излучения с веществом в ультрафиолетовой (УФ), видимой, инфракрасной (ИК) областях спектра.
Энергию, которой обладает молекула, можно представить как сумму энергий внешних валентных электронов Ее, энергии колебательного Еколеб и вращательного Евращ движения электронов в молекуле:
![]() ,
,
причем по абсолютной
величине
![]() .
.
Молекула имеет квантованные энергетические уровни, поэтому возможны только такие переходы молекул с одного энергетического уровня на другой, энергия которых подчиняется закону:
![]() ,
,
где ΔЕ энергия перехода молекулы из основного состояния в возбужденное, h постоянная Планка, равная 6,63·1034 Дж с, с скорость света, равная 3·108 м/с, λ длина волны поглощенного электромагнитного излучения, м, частота этого излучения, с1.
Поглощение энергии, соответствующей длине волны , вызывает скачкообразное изменение энергии состояния молекулы. При облучении образца излучением с непрерывно меняющейся частотой часть излучения из определенных участков спектра поглощается молекулой. Регистрируя интенсивность прошедшего излучения в зависимости от длин волн или волновых чисел, получают спектры поглощения.
Как правило, спектры представляются в виде зависимости интенсивности прошедшего излучения I или пропускания T=I/I0 (отношение интенсивности прошедшего излучения к интенсивности падающего излучения, выраженное в процентах) или оптической плотности A=lg(I0/I) от частоты падающего излучения ν или длины волны λ или волнового числа (величина, равная 1/λ, выражается обычно в см1).
Изменениям энергетического состояния внешних валентных электронов соответствует поглощение энергии в ультрафиолетовой (λ=180 - 400 нм) и видимой (λ=400-800 нм) области спектра. Валентные и деформационные колебания атомов в молекуле и изменения вращательной энергии отражаются в ИК-спектрах. Инфракрасная область спектра подразделяется на дальнюю (λ=20-200 мк), среднюю (λ=2-20 мк) и ближнюю (λ=0.8-2 мк) области. Чаще всего используют спектры, снятые в средней ИК-области.
Типовая схема спектрофотометра представлена на рис. 6.1.
|
|
|
Рис. 6.1. Блок-схема спектрофотометра: 1 источник излучения; 2 кювета с пробой; 3 кювета сравнения; 4 монохроматор; 5 детектор; 6 регистрирующее устройство |

Источником излучения может служить дейтериевая лампа (в УФ-спектроскопии), лампа накаливания с вольфрамовой нитью (спектроскопия в видимой области) или стержень, нагретый до температуры порядка 400С, излучающий в ИК-области.
Монохроматор – оптический прибор, позволяющий выделять излучение заданной длины волны (обычно с точностью до 1 нм) или проводить сканирование по длинам волн в выбранном диапазоне.
Детектором для спектрометров, работающих в видимой и УФ-области, обычно служит ФЭУ или диодная матрица, для ИК-спектрометров – термопара, которая регистрирует изменения температуры, возникающие в результате поглощения части ИК- (теплового) излучения.
Регистрирующим устройством является компьютер или самописец.
Излучение от соответствующего аналитическому методу источника направляется на две кюветы, в одной из которых находится анализируемая проба, обычно растворенная в каком-либо растворителе, а в другой раствор сравнения, т.е. тот же растворитель, но без анализируемой пробы. Детектор сравнивает интенсивность излучений, прошедших через две кюветы. Полученная разность интенсивностей характеризует природу анализируемого вещества.
В УФ- и видимой спектроскопии анализируемые образцы представляют собой жидкие вещества или растворы. Используемый растворитель должен быть прозрачным в исследуемом диапазоне, чтобы не мешать определению изучаемого вещества.
Для анализа методом ИК-спектроскопии можно использовать более разнообразные образцы. Они могут представлять собой газы, закачанные в герметичные кварцевые кюветы, растворы в четыреххлористом углероде, сероуглероде или других растворителях (прозрачных в изучаемом диапазоне длин волн), тонкие пленки жидких веществ, порошок твердого вещества, растертый в вазелиновом масле. Чаще всего используются пробы твердых веществ, запрессованных в таблетки бромида калия, хлорида натрия и т.п. солей, потому что ИК-спектры неорганических солей и органических веществ практически не накладываются друг на друга. Анализируемые образцы для метода многократно нарушенного полного внутреннего отражения (МНПВО) представляют собой зеркально отполированные срезы твердого вещества.
Более современными приборами являются ИК-Фурье спектрометры, в которых спектр получают в результате Фурье-преобразования интерферограммы исследуемого излучения. Схема ИК-Фурье спектрометра представлена на рис. 6.2.
|
|
|
Рис. 6.2. Оптическая схема ИК-Фурье спектрометра: 1 – неподвижное зеркало интерферометра; 2 – подвижное зеркало; 3 – светоделительная пластина; 4 – источник излучения; 5 – исследуемый образец; 6 – детектор |

Светоделительная пластина направляет часть светового потока на неподвижное зеркало, а другую часть на подвижное (которое непрерывно колеблется между двумя положениями), в результате две части потока проходят различное расстояние. В результате наложения этих частей потока друг на друга получается интерферограмма, вид которой зависит от оптической разности хода лучей и представляет собой Фурье-образ спектра, т.е. функции распределения энергии излучения по частотам.
При поглощении образцом энергии с какой-либо частотой наблюдается уменьшение интенсивности интерферограммы, соответствующей этой частоте. После проведения Фурье-преобразования в полученном спектре наблюдается полоса поглощения образца. Преобразование Фурье:
![]()
осуществляется на ЭВМ.
Использование
Фурье-спектрометров имеет несколько
преимуществ. Выигрыш Фелджета, или
мультиплекс-фактор, связан с тем, что
любая точка интерферограммы содержит
информацию о всей исследуемой области
спектра. На детектор в каждый момент
поступают сигналы, соответствующие
всем частотам. За одно сканирование (за
время t1) регистрируется
спектр с таким же отношением сигнал/шум
![]() ,
как и для дисперсионного спектрометра
за время t2 (на
несколько порядков большее, чем t1
). Если для получения спектра на
Фурье-спектрометре затратить время t2,
то соотношение сигнал/шум возрастет в
соответствии с уравнением:
,
как и для дисперсионного спектрометра
за время t2 (на
несколько порядков большее, чем t1
). Если для получения спектра на
Фурье-спектрометре затратить время t2,
то соотношение сигнал/шум возрастет в
соответствии с уравнением:
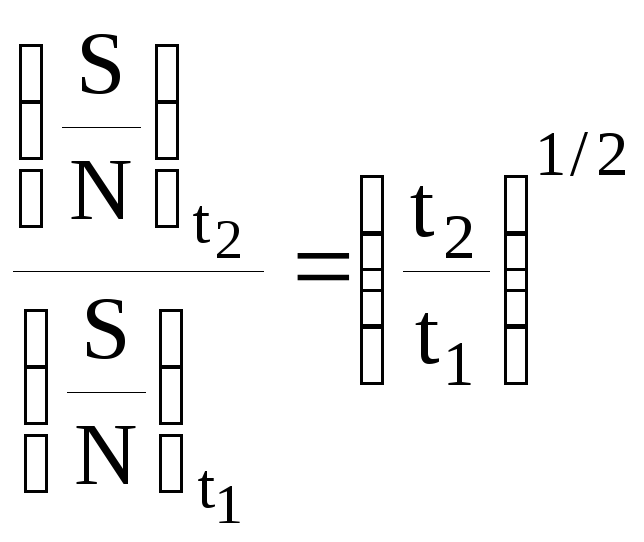 .
.
Другое важное преимущество – геометрический фактор – определяется отсутствием в Фурье-спектрометре щелей, что дает выигрыш в светосиле в 100–200 раз. Это позволяет уменьшить время регистрации спектров, повысить разрешающую способность приборов, уменьшить их габариты.
Оптическая спектроскопия изучает спектры поглощения излучений. Вид спектров является индивидуальным для данного вещества, что позволяет использовать их для качественного анализа. Причем наиболее информативными в этом смысле являются спектры инфракрасного диапазона.
Оптические спектральные методы можно использовать также для количественного анализа веществ. Количественные зависимости определяются законом Ламберта-Бугера-Бера:
![]() ,
,
где А – оптическая плотность (абсорбция) поглощающей среды; I0 – интенсивность падающего излучения; I – интенсивность излучения, прошедшего через поглощающий слой толщины l; - коэффициент экстинции (поглощения, абсорбции), зависящий от длины волны излучения и природы вещества; с – концентрация определяемого элемента в поглощающем слое.
Данный закон не всегда соблюдается абсолютно точно. Отклонения могут возникать из-за немонохроматичности излучения. Поскольку коэффициент экстинции зависит не только от природы вещества, но и от длины волны, то в случае недостаточно узкого диапазона длин волн, используемого при количественном измерении, возникает неоднозначность в определении значения . Высокие концентрации определяемых веществ также могут привести к погрешности в измерениях из-за влияния межмолекулярных взаимодействий, образования внутримолекулярных водородных связей.
Закон Ламберта-Бугера-Бера обладает свойством аддитивности, т.е.
A =icil,
где A - оптическая плотность смеси, i - молярный коэффициент поглощения i-го вещества, ci – концентрация i-го вещества, l – толщина поглощающего слоя смеси веществ.
Пользуясь аддитивностью закона Ламберта-Бугера-Бера можно проводить определение концентрации веществ в смеси без ее разделения. Для этого требуется знать какие вещества находятся в анализируемом образце и иметь справочные данные о зависимости их коэффициентов экстинции от длины волны. Проведя измерение оптической плотности образца при двух различных длинах волн и решив систему из двух уравнений, можно рассчитать неизвестные концентрации двух веществ, находящихся в смеси, без ее разделения:
1: A1 = l (1ici +1jcj)
2: A2 = l (2ici +2jcj).
Если в смеси имеются три вещества, оптическую плотность следует определять при трех различных длинах волн, и решать, соответственно, систему из трех уравнений и т.д.
Электронные спектры, т.е. поглощение энергии излучения электронами вещества, наблюдаются при длинах волн, соответствующих УФ и видимой области. Электронное поглощение связано с переходами валентных электронов кратных связей, электронов неподеленных пар гетероатомов и электронов сопряженных -систем. Эти электроны имеют различную энергию и, следовательно, возбуждаются излучением с различными длинами волн.
Электронные спектры поглощения чистых веществ обычно состоят из нескольких широких полос и не имеют узких пиков, поскольку любой электронный переход сопровождается теми или иными изменениями во вращательных или колебательных состояниях молекул. Важнейшей характеристикой полос является длина волны, при которой наблюдается максимум поглощения (max). Иногда, если соседние полосы частично перекрываются, в качестве характеристики спектра указывают и минимум поглощения. При более сильном перекрывании полос одна из них может проявляться в спектре только в виде плеча. Обычно в таких случаях измеряют длину волны, соответствующую точке перегиба.
Чем больше сопряженных связей имеется в молекуле, тем дальше длина волны, соответствующая максимуму поглощения, сдвинута в сторону длинных волн. Например, для этилена max составляет 162 нм, для бутадиена – 217 нм, а для бензола – 255 нм. Введение алкильных групп в бензольное ядро вызывает сдвиг max бензола в длинноволновую область спектра. Для алкилнафталинов max = 275-290 нм, для алкилпроизводных антрацена и фенантрена характерны широкие полосы поглощения с несколькими максимумами, соответственно в области 310-380 нм и 280-310 нм.
В случае гетероатомных соединений (кислородные, сернистые, азотистые соединения) алифатического ряда полосы поглощения находятся в коротковолновой области УФ-спектра ( 200 нм). Если гетероатом находится рядом с двойной связью или ароматическим ядром, то поглощение происходит в области 200-300 нм.
Электронные спектры используются для изучения экстрактов, различных фракций, полученных из угля деструктивными методами, жидких продуктов, выделенных из нефти и т.п.
Спектры смесей имеют вид сплошных кривых, отличающихся уровнем интенсивности и наклоном. С качественной точки зрения электронные спектры смесей малоинформативны.
Спектроскопия в УФ- и видимой области широко используется для количественного определения концентраций веществ (в качестве детекторов в хроматографии).
Если в смеси имеются три вещества, оптическую плотность следует определять при трех различных длинах волн, и решать, соответственно, систему из трех уравнений и т.д.
ИК-спектроскопия является важным методом установления структуры молекул, этот метод часто используется в качественном анализе веществ. Каждой функциональной группе, группе атомов и связи в молекуле исследуемого вещества соответствуют несколько полос спектра, отвечающих частотам поглощенных лучей (рис. 6.3).

4000 3800 2800 2000 1800 1200 800
1/ (cm-1)
Рис. 6.3. ИК-спектр 1,3,5-триметилбензола
Сложное колебательное движение молекулы всегда можно представить в виде суммы относительно простых составляющих, каждый тип которых характеризуется определенной частотой (фундаментальной). Колебания разделяются на валентные и деформационные. Деформационные колебания состоят в изменении величин валентных и двугранных углов, а валентные колебания в изменении длин связей и могут быть симметричными и асимметричными.
Сумма или разность двух различных фундаментальных частот называются составными частотами, а частоты, кратные каким-либо фундаментальным частотам, – обертонами.
В ИК-спектрах активны колебания, связанные с изменением дипольного момента. В асимметричных молекулах любое колебание является активным. Только симметричные молекулы могут совершать колебания, при которых изменение дипольного момента точно равно нулю, и поэтому такие колебания являются неактивными в ИК спектрах. Например, в молекуле СО2 возможны деформационные колебания, симметричные валентные колебания и асимметричные валентные колебания, но в ИК спектре этой молекулы полоса, частота которой соответствует симметричному валентному колебанию не обнаруживается, поскольку при таком колебании дипольный момент не меняется.
Частоты колебания связей чувствительны к их окружению в молекуле, особенно в тех случаях, когда в окружении имеются полярные группы. Атомы, образующие связь, могут вступать еще в донорно-акцепторные межмолекулярные связи, ароматические кольца могут иметь один или несколько заместителей (особенно полярных). Поэтому в таблицах характеристических частот вместо одного значения указывают границы полос поглощения.
Достаточно интенсивные полосы поглощения, проявляющиеся в области, характерной для определенной группы атомов, и пригодные для идентификации этой группы, называются характеристическими полосами поглощения или характеристическими частотами.
Положение полосы определяется силой связи и массой связываемых атомов. Чем сильнее связь и чем меньше массы атомов, тем выше частота, при которой данная связь поглощает излучение, т.е. тем больше энергии нужно затратить на колебание связи. Так, например, сила связи возрастает при переходе от одинарной к двойной и тройной связям, и соответственно возрастают волновые числа валентных колебаний от 700-1500 см1 до 1600-1800 и до 2000-2500 см1. Частота валентного колебания связи O – H, равна 3600 см1, но она снижается до 2630 см1 для связи O – D, у которой сила связи та же самая, а масса одного из атомов (дейтерия) больше.
Поскольку поглощение имеет место только тогда, когда колебание приводит к изменению распределения заряда внутри молекулы, то чем больше это изменение, тем сильнее поглощение. Соответственно полосы в спектре углеводородов, состоящих только из атомов углерода и водорода, являются слабыми, а полосы, относящиеся к связям, соединяющим атомы, сильно различающиеся между собой по степени электроотрицательности, например C N, C – O, C = O, обычно довольно сильными. Частоты деформационных и валентных колебаний одинарных связей расположены в одной и той же области спектра, и тем не менее полосы, соответствующие валентным колебаниям C – N и C – O, могут быть обнаружены достаточно легко, так как они сильнее, чем полосы валентных колебаний связи C – C.
Кратные связи сильнее одинарных, а связи типа X – H (N – H, O – H, C – H и т.д.) имеют очень легкий концевой атом водорода. Колебания такого рода связей испытывают лишь незначительные воздействия со стороны остальной части молекулы. Таким образом, валентные колебания этих специфических связей проявляются в диапазоне 3600-1500 см1.
В области ниже 1600 см1 лежат полосы поглощения, обусловленные растяжением одинарных связей, а также деформацией угла между связями. Одинарные связи обычно соединены кумулятивно, например С – С – О. Это приводит к более сильному взаимодействию между связями, расширению области проявления соответствующих полос поглощения и большой чувствительности положения полос даже к небольшим изменениям структуры.
Низкочастотная область инфракрасного спектра (1300-650 см1) известна как область «отпечатков пальцев» каждое соединение имеет в этом интервале свою специфическую спектральную кривую, по которой можно точно идентифицировать соединения.
Спектры строятся в координатах интенсивность I – частота (длина волны , волновое число 1/). Интегральная интенсивность (площадь под кривой) пика пропорциональна количеству связей с частотой колебания, соответствующей данному пику. Для расшифровки спектров используют литературные данные по значениям характеристических частот валентных и деформационных колебаний отдельных типов связей, атомных и функциональных групп.
В ИК-спектроскопии не слишком хорошо соблюдаются количественные зависимости, поскольку большинство полос спектра плохо разделяются с соседними полосами. Тем не менее при анализе структур природных энергоносителей часто пользуются полуколичественным (приближенным) расчетом структурных параметров Ki:
![]() ,
,
где Ki - структурный параметр i-й полосы, Ii - интенсивность i-й полосы, а Iстанд - интенсивность стандартной полосы.
Чаще всего в качестве стандарта выбирают полосу деформационных колебаний СН2-группы, так как эта группа входит в большинство органических соединений.
Для анализа методом ИК-спектроскопии твердых непрозрачных проб используется специфический метод многократно нарушенного полного внутреннего отражения (МНПВО), основанный на отражении пучка излучения от поверхности раздела между средами с различным коэффициентом преломления (рис. 6. 4).
Интенсивность падающего излучения I0 и выходящего из ячейки излучения I неодинаковы из-за того, что часть излучения поглощается полированной поверхностью пробы, причем спектр этого поглощенного излучения определяется природой образца.
|
|
|
|---|---|
|
Рис. 6.4. Кювета, используемая в методе многократного нарушенного полного внутреннего отражения |
|

Данные ИК-спектров углей и продуктов их переработки используются для идентификации структур. Однако результаты таких исследований в углехимии носят скорее качественный характер и не могут претендовать на количественное определение каких-либо структурных фрагментов. Дело в том, что угольное вещество неоднородно и состоит из множества органических соединений, отличающихся по структуре, количеству влаги и минеральных включений, находящихся в сложном взаимодействии. Поэтому в отличие от простых соединений спектры угольного вещества состоят из размытых полос, которые включают в себя множество частот и однозначное отнесение их к конкретным характеристическим частотам проблематично.
При изучении продуктов переработки углей обычно рассматривают соотношения, например, интенсивностей характеристических полос С – Салиф и С = С, распределение кислорода по различным функциональным группам (карбонильным 1690 см1 и гидроксильным, особенно связанным водородной связью, 3100 см1). По изменению интенсивности смеси двух веществ по сравнению с расчетной суммой можно обнаружить наличие химического взаимодействия. При анализе продуктов переработки нефти чаще всего сначала проводится разделение смеси на достаточно узкие фракции, которые затем анализируются.
Для количественного анализа ИК-спектрометры могут использоваться в хроматографии. Главным их достоинством является универсальность (подходят для всех веществ). При этом возможно вести регистрацию только отдельных выбранных классов веществ. Например, при рабочей длине волны, соответствующей деформационным колебаниям СН2-группы, можно определять все органические вещества, а если выбрать в качестве рабочей длину волны, соответствующую поглощению карбонильной группы, то можно определять только карбонилсодержащие соединения.