

1.7. Зависимость теплового эффекта от температуры
Тепловой эффект процесса слабо зависит от давления и может существенно изменяться с температурой.
![]() и
и![]() .
.
Эти термодинамические соотношения определяют температурную зависимость теплового эффекта химических реакций и являются дифференциальными выражениеми закона Кирхгофа, одна из формулировок которого гласит:
Температурный коэффициент теплового эффекта равен изменению теплоемкости системы (или разности сумм теплоемкостей продуктов реакции и исходных веществ), происходящему в результате процесса.
Согласно представленным уравнениям,
влияние температуры на тепловой эффект
обуславливается знаками величин
![]() и
и
![]() .
.
Проведем анализ зависимости (рис.4.):
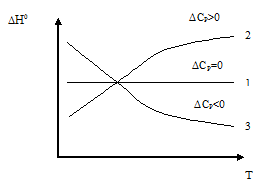
 ,
, ,
таким образом, тепловой эффект процесса
не зависит от температуры
,
таким образом, тепловой эффект процесса
не зависит от температуры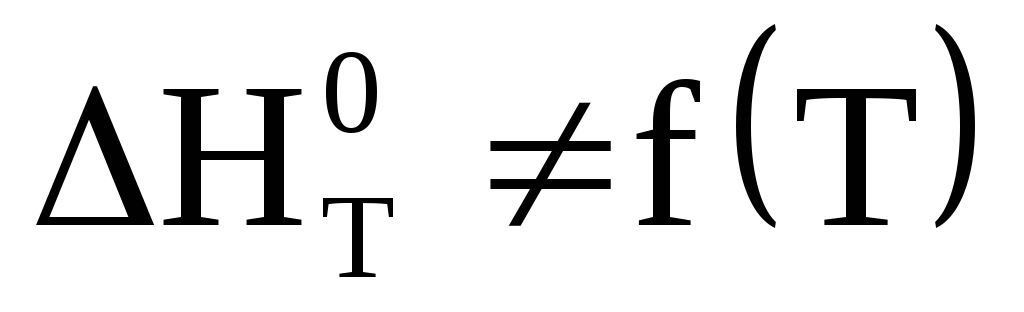 ;
; ,
в этом случае тепловой эффект процесса
с ростом температуры увеличивается;
,
в этом случае тепловой эффект процесса
с ростом температуры увеличивается; ,
в этом случае тепловой эффект процесса
с ростом температуры уменьшается.
,
в этом случае тепловой эффект процесса
с ростом температуры уменьшается.
Однако, нам необходимо знать каков тепловой эффект процесса при данной, конкретной температуре.
Интегрируя уравнения Кирхгофа
![]() и
и![]() ,
получим выражения для расчета тепловых
эффектов реакций при произвольных
температурах:
,
получим выражения для расчета тепловых
эффектов реакций при произвольных
температурах:
V = const:
![]()
Р = const:
![]() интегральная форма закона Кирхгофа
интегральная форма закона Кирхгофа
где
![]() ,
,![]() – тепловые эффекты, осуществляемые в
стандартных условиях при V,Р = const, которые
рассчитываются по закону Гесса с
использованием стандартных теплот
образования веществ.
– тепловые эффекты, осуществляемые в
стандартных условиях при V,Р = const, которые
рассчитываются по закону Гесса с
использованием стандартных теплот
образования веществ.
Если, например,
![]() и величина отрицательная, то графически
зависимость теплового эффекта от
температуры имеет вид (рис.5.):
и величина отрицательная, то графически
зависимость теплового эффекта от
температуры имеет вид (рис.5.):
В общем случае тепловой эффект при
постоянной теплоемкости можно определить
из уравнения:
общем случае тепловой эффект при
постоянной теплоемкости можно определить
из уравнения:
![]()
Часто при проведении расчетов возникает необходимость учета температурной зависимости теплоемкости компонентов реакции. Как правило, аналитическая форма функциональной зависимости C = f(T)неизвестна.В этом случае обычно используют эмпирические соотношения в виде степенных рядов для отдельных компонентов реакции С = + Т + Тn, а затем уже рассчитывается температурная зависимость теплоемкости химической реакции. Тогда, зависимость теплоемкости от температуры приводится к виду
![]() ,
где
,
где![]() и т.д.
и т.д.
Подставляя это выражение в уравнения Кирхгофа, для стандартных условий, получим
![]() .
.
После интегрирования уравнение приводятся к виду:
![]() .
.
Если в рассматриваемом интервале температур имеют место фазовые переходы "твердое тело – жидкость", "жидкость – пар", то в уравнениях температурной зависимости теплового эффекта появляются дополнительные составляющие, учитывающие тепловые эффекты фазовых переходов:
 .
.
– изменения изохорной и изобарной теплоемкостей в соответствующих фазовых состояниях (твердом, жидком и газообразном). Температурная зависимость энтальпии, с учетом фазовых переходов, качественно показана на рис.6.

По величине
![]() воспользовавшись соотношением Клаузиуса,
можно рассчитать тепловой эффект
процесса в изохорических условиях, т.е.
приV=const:
воспользовавшись соотношением Клаузиуса,
можно рассчитать тепловой эффект
процесса в изохорических условиях, т.е.
приV=const:![]() ,
гдеn – разность
между числом молей газообразных веществ
в конечном и начальном состояниях
,
гдеn – разность
между числом молей газообразных веществ
в конечном и начальном состояниях![]() .
(например, СО2+С=2СО;n=2-1=1.
.
(например, СО2+С=2СО;n=2-1=1.
При n=0,![]() .
.
2. Второе начало термодинамики. Энтропия.
Все процессы, происходящие в природе, отличаются определенной направленностью, т.е. они совершаются сами собой только в одном направлении, хотя первый закон не запрещает их протекания в обратном направлении.
Необходимо ввести критерии, позволяющие определить возможность и наиболее вероятное направление, а также пределы протекания самопроизвольных процессов.
Попытки найти такой критерий привели к предположению, что самопроизвольно осуществляются только экзотермические процессы. Однако, мы знаем, что многие спонтанные термодинамические процессы, в частности химические реакции, могут протекать как с поглощением теплоты
![]()
![]()
так и с ее выделение:
![]()
![]()
сопровождаясь возрастанием неупорядоченности систем (газ более разупорядочен, чем кристалл или жидкость).
Более того, многие реакции, например,
![]()
![]()
не протекают самопроизвольно, однако сопровождаются отрицательным изменением энтальпии. Поэтому – изменение внутренней энергии или энтальпии не может служить критерием, позволяющим предвидеть направление протекания процесса (реакции).
Критерии возможности направления протекания процесса определяет II закон термодинамики, который был сформулирован в 60-х годах прошлого столетия.
С его помощью решаются сформулированные выше два круга вопросов.
определяется возможность, направление и предел протекания термодинамического процесса в данной системе в заданных условиях.
устанавливаются параметры протекания термодинамического процесса в заданном направлении с необходимой полнотой.
Клаузиусом была введена функция, названная энтропией S, и сформулировано два принципа: принцип существования энтропии и принцип возрастания энтропии в изолированных системах.
Принцип существования энтропии
Для каждой термодинамической системы существует функция состояния системы S - энтропия, определяемая тем, что ее дифференциал (dS) при элементарном обратимом (равновесном) процессе равен отношению бесконечно малого количества теплоты (q), сообщенного системе, к абсолютной температуре системы:
![]() .
.
Принцип возрастания энтропии
Энтропия изолированной системы при наличии в ней необратимых процессов всегда возрастает
![]() .
.
Таким образом, в изолированной системе обратимые процессы протекают без изменения энтропии, а необратимые - с возрастанием энтропии.
Свойства энтропии
Энтропия– функция состояния системы. Ее изменение зависит от начального и конечного состояний системы.
Энтропияявляется мерой
неупорядоченности системы.![]() .
.
Энтропия – аддитивная функция, т.е. энтропия системы равна сумме энтропий ее частей (это экстенсивный параметр!)
Энтропия– вероятностная функция. Она показывает вероятность процесса и направление наиболее его вероятного протекания:
![]() ,
(уравнение Больцмана)
,
(уравнение Больцмана)
где W- термодинамическая вероятность;k– постоянная Больцмана. Термодинамическая вероятность – это число микросостояний, т.н. вариантов в расположении частиц (атомов молекул), которыми может быть осуществлено данное макросостояние.
Термодинамическую вероятность, которая может принимать очень большие значения, следует отличать от математической, всегда меньшей единицы.
Энтропия – мера рассеивания энергии в окружающую среду, т.е. та часть энергии, которая в полезную работу не превращается!!!
Все изложенное выше находит отражение в формулировках второго закона термодинамики:
1.Тепло не может самопроизвольно переходить от более холодного к более теплому телу (Клаузиус).
2.Невозможно создать вечный двигатель второго рода, т.е. такую тепловую машину, в которой бы теплота полностью переходила в работу.
3.Любая система, представленная сама себе, будет изменяться в направлении состояния, отвечающего максимальной вероятности (Льюс).
4. Энтропия изолированной системы увеличивается или остается постоянной
![]()
> для самопроизвольных необратимых процессов
= для обратимых процессов.
В последней четвертой формулировке заключен критерий самопроизвольности процессов в изолированных системах: спонтанно в такой системе могут протекать те процессы, которые сопровождаются увеличением энтропии (перемешивание газов, выравнивание температур и давлений).
расчет изменения энтропии в обратимых процессах
(равновесных процессах)
1. Нагревание веществ от Т1 до Т2 (неизотермический процесс)
Уравнения для вычисления S изобарного (P=const) и изохорного (V=const) нагревания 1 моля веществ имеют вид:
![]() и
и
![]() .
.
Для
n
молей:
![]() и
и![]() .
.
2. Изотермическое расширение газа, объем которого изменяется при некоторой температуре Т от V1 до V2:
![]() .
.
Для
n
молей:
![]()
3. Изменение энтропии фазовых переходов вычисляется по уравнению
![]() ,
,
где
![]() - теплота фазового перехода,
- теплота фазового перехода,![]() -
температура фазового перехода.
-
температура фазового перехода.
Например, теплота фазового перехода воды из жидкого состояния в парообразное (процесс испарения) составляет около 41 кДж/моль. Следовательно, изменение энтропии составляет:
![]() Дж/моль·К.
Дж/моль·К.
4. Изменение энтропии химических реакций, протекающих в изотермических условиях, вычисляется как разность сумм энтропий конечных и исходных веществ участвующих в реакции
![]() .
.
![]() ,
,
где SC, SД, SA, SB - абсолютные энтропии веществ - участников реакции; а, в, с, д - стехиометрические коэффициенты.
Абсолютные значения энтропий веществ вычислены для стандартных условий протекания реакций и представлены в термодинамических справочниках.
Определение абсолютных энтропий веществ стало возможным после формулировки постулата Планка, который позволил определить начало отсчета энтропии - состояния с минимальной энтропией при абсолютном нуле температуры.
Постулат Планка составляет содержание 3 - го закона термодинамики и формулируется так:
«Энтропия полностью упорядоченных кристаллических тел при температуре абсолютного нуля равна 0».
S0К
=0, тогда
![]() .
.
Стандартная энтропия индивидуальных веществ (So298) имеют размерность Дж/мольК, т.е. это количество теплоты, которое рассеивается в окружающую среду, если моль вещества нагревается на 1о.
Т. е. Энтропия индивидуальных веществ характеризует энергетически бесполезную теплоту; ту теплоту, которая в работу не переходит.
При расчетах S системы следует не забывать, что энтропии веществ - величины экстенсивные, т.е. зависящие от массы.
применение II закона термодинамика для неизолированных систем
Многие процессы химической технологии
протекают при постоянном давлении и
температуре, если они проводятся в
открытых аппаратах, илипри постоянном
объеме и температуре, если они
проводятся в закрытых аппаратах,
например, в автоклавах. В химической
технологии о направлении процесса и
равновесии в системе при постоянных
давлении и температуре судят по энергии
Гиббса или при постоянных объеме и
температуре — по энергии Гельмгольца.
Это связано с тем, что на практике
рассмотрение изолированных систем и
использование
![]() в качестве критерия равновесия и
направления процесса крайне неудобно,
так как рассмотрение изолированной
системы часто встречает большие
затруднения.
в качестве критерия равновесия и
направления процесса крайне неудобно,
так как рассмотрение изолированной
системы часто встречает большие
затруднения.
Изохорно-изотермические условия (V=const, T=const)
по уравнению Гиббса - Гельмгольца
![]() .
.
При
![]() - самопроизвольный процесс возможен в
прямом направлении; при
- самопроизвольный процесс возможен в
прямом направлении; при![]() система находится в состоянии равновесия;
при
система находится в состоянии равновесия;
при![]() самопроизвольный процесс в прямом
направлении невозможен, возможен
обратный процесс.Таким
образом, если в системе, находящейся
при Т и V = const, происходит какой-либо
неравновесный самопроизвольный процесс,
то энергия Гельмгольца уменьшается до
тех пор, пока система не достигнет
равновесия.
самопроизвольный процесс в прямом
направлении невозможен, возможен
обратный процесс.Таким
образом, если в системе, находящейся
при Т и V = const, происходит какой-либо
неравновесный самопроизвольный процесс,
то энергия Гельмгольца уменьшается до
тех пор, пока система не достигнет
равновесия.
Изобарно-изотермические условия (Р=const, T=const)
по уравнению Гиббса - Гельмгольца
![]() .
.
При
![]() - самопроизвольный процесс возможен в
прямом направлении; при
- самопроизвольный процесс возможен в
прямом направлении; при![]() система находится в состоянии равновесия;
при
система находится в состоянии равновесия;
при![]() самопроизвольный процесс в прямом
направлении невозможен, возможен
обратный процесс.Таким
образом, если в системе, находящейся
при Т и V = const, происходит какой-либо
неравновесный самопроизвольный процесс,
то энергия Гельмгольца уменьшается до
тех пор, пока система не достигнет
равновесия.
самопроизвольный процесс в прямом
направлении невозможен, возможен
обратный процесс.Таким
образом, если в системе, находящейся
при Т и V = const, происходит какой-либо
неравновесный самопроизвольный процесс,
то энергия Гельмгольца уменьшается до
тех пор, пока система не достигнет
равновесия.