

Биофизика 02
.pdfтропоколаген.
Іншим типом конформацій є βFформа (рис. 3.16). Така структура утворюється сукупністю декількох поліпептидних ланцюгів (рис. 3.17) або з одного ланцюга, декілька разів вигнутого (кросFβFформа) (рис. 3.18) і стабілізується водневими зв’язками, що виникають між поруч розташованими ланцюгами. У результаті утворюється структура типу складчастого шару. Окремі ланцюги в такій структурі можуть розташовуватися паралельно й антипаралельно. У паралельній βFформі кути ϕ і ψ складають 61° і 239°, а в антипаралельній — 380° і 325°, відповідно.
Убілках зустрічаються і неупорядковані ділянки, на яких кути
ϕі ψ мають значення, відмінні від тих, що були зазначені вище. Частка неупорядкованих ділянок у деяких білках може склада-
ти до 50–60%. Так, наприклад, у гемоглобіні 75% поліпептидних ланцюгів знаходяться у вигляді αFспіралі, а залишені 25% являють собою неупорядковані ділянки. Останні забезпечують вигини лан-
цюгів у просторі, зокрема, такі ділянки знаходяться в місці вигину кросFβFформи.
Імовірність зустріти ту або іншу амінокислоту в αF, βFформах або неупорядкованих ділянках різна. Так, наприклад, у αFспіралях частіше
усього зустрічаються такі амінокислоти як глутамінова кислота, аланін, лейцин, у βFформах — метіонін, валін, ізолейцин, у неупорядкованих ділянках — гліцин і пролін (останній не здатний утворювати водневих зв’язків, тому що насправді є
не аміноF, а імінокислотою). Знаючи частоту зустрічаємості амінокислот у різних видах конформацій білка, можна на підставі інформації про первинну структуру з деякою імовірністю (до 70%) передбачити вторинну.
Поліпептидний ланцюг стабілізується в просторі не тільки водневими зв’язками, але і гідрофобними взаємодіями, іонними і дисульфідними (–S–S–) зв’язками, останні утворюються між амінокислотними залишками далеко віддаленими один від одного в ланцюзі. У результаті цих взаємодій білковий ланцюг виявляється складеним в деяку компактну структуру, у якій чергуються упорядковані і неупорядковані ділянки (третинна структура білка).
Деякі білкові молекули містять у своєму складі не одну, а кілька поліпептидних ланцюгів (субодиниць). Кожний ланцюг має свою третинну структуру і зв’язаний з іншими ланцюгами нековалентними
Рис. 3.15. Розташування водневих зв’язків у α[спіралі
білка.
61

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис. 3.17. β[шар із кількох поліпеп- |
Рис. 3.18. Крос[β[форма. |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Рис. 3.16. Розта- |
||||||||||||||||||||||||
шування водневих |
тидних ланцюгів. |
|
|
|
|
|
|
|
|
|
||||||||||||||
зв’язків у β[формі. |
|
|
|
|
|
|
|
|
|
|
||||||||||||||
зв’язками, формуючи четвертинну структуру. Білки, що володіють четвертинною структурою, мають у своєму складі точно певне число субодиниць, наприклад, у гемоглобіні їх чотири.
Зв’язки, що стабілізують вторинну, третинну і четвертинну структури, є слабкими (крім ковалентних дисульфідних). Тому зміна умов навколишнього середовища може привести до їхнього розриву й утворення нових зв’язків. Утворюється нова конформація, енергетично вигідна в даних умовах, тобто відбувається конформаційний перехід.
Серед амінокислот, що входять до складу білків, є як гідрофільні (аргінін, аспарагін, гістидин, глутамін, лізин, серин, тирозин і треонін), так і гідрофобні (інші 12). До складу того самого білка входять, як правило, і ті, і інші амінокислоти. Так як білкова молекула є рухливою, то вона у воді (полярному розчиннику) прагне згорнутися таким чином, щоб її гідрофільні амінокислоти контактували з ним, а гідрофобні були “заховані” усередині. У результаті усередині молекули утворюється гідрофобне ядро, покрите зверху гідрофільною оболонкою. Така структура називається білковою глобулою (від лат. globulus — кулька). Її утворення забезпечує формування компактної структури при великій молекулярній масі.
Основну роль у формуванні білкової глобули грають саме гідрофобні взаємодії, які сприяють виштовхуванню неполярних амінокислот із водяного середовища, а не виграш енергії при утворенні водневих зв’язків між полярними амінокислотами і водою, тому що приблизно така ж кількість енергії виділяється і при формуванні водневих зв’язків між амінокислотами в білку.
Форма глобули визначається співвідношенням числа полярних і неполярних амінокислотних залишків. Якщо в білку b = bs (де b — відношення числа полярних залишків до неполярних, bs — відношення об’єму гідрофільної фази до об’єму гідрофобного
62
ядра), то глобула буде прагнути утворити сферу. При b > bs, тобто полярних залишків більше, ніж необхідно, щоб покрити гідрофобне ядро, глобула прийме витягнуту форму. При b < bs полярних залишків не вистачає і частині гідрофобного ядра приходиться контактувати з водою. Щоб уникнути цього кілька таких молекул утворюють комплекс одна з однією. На рисунку 3.19 наведені можливі форми білкових глобул у залежності від параметра b.
§ 18. Ферментний каталіз
Однією з основних функцій білків є ферментативна. БілкиFферменти здатні прискорювати біохімічні реакції в 108–1010 разів у порівнянні з тим, якби ці реакції відбувалися без участі ферментів.
Відповідно до формули Арреніуса, константа швидкості реакції дорівнює:
(3.5) де А — константа, що визначає частоту зіткнень молекул, які реагують; Еакт — енергія активації — висота потенційного бар’єра, який повинна перебороти система для здійснення реакції; R — універсальна газова
стала; T — температура; множник 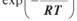 визначає долю молекул, енергія яких перевищує енергію активації.
визначає долю молекул, енергія яких перевищує енергію активації.
Ферменти ніколи не зрушують хімічної рівноваги в реакціях, обумовленої різницею вільних енергій продукту і субстрату. Ці реакції відбуваються й у відсутності ферменту, але з набагато меншою швидкістю. Роль ферментів зводиться до зменшення енергії активації даної реакції, а, отже, відповідно до (3.5), — збільшення константи швидкості.
Ферменти мають високу специфічність і, як правило, каталізують тільки певні реакції або реакції за участю вузького класу сполук. Пер-
Гідрофільна оболонка
Гідрофобне ядро
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
а |
б |
в |
|
|
||
Рис. 3.19. Різні форми білкової глобули: а) сфера (b = bs ); б) еліпсоїд (b > bs ); в) надмолекулярні структури (b < bs ).
63

шою моделлю, що пояснювала специфічність ферменту, стала модель Фішера, відповідно до якої субстрат стерично відповідає активному центру ферменту (ділянці ферменту, до якої приєднується субстрат). Ця модель одержала назву ключFзамок. Відповідно до більш пізньої моделі Кошланда (моделі індукованої відповідності), приєднання певного субстрату викликає конформаційні перебудови у ферменті, у результаті чого його каталітичні групи орієнтуються в просторі таким чином, що виявляються здатними здійснити перетворення субстрату в продукт. Ця модель пояснює той факт, що приєднання до ферменту деяких речовин, структурно схожих на субстрат, не приводить до їхніх хімічних перетворень.
Розглянемо кінетику ферментативних реакцій. У найпростішому випадку приєднання субстрату до вільного ферменту Е0 приводить до утворення ферментFсубстратного комплексу [ES] із константою швидкості реакції k1. Із комплексу або утворюється продукт Р (константа k2) або комплекс розпадається на субстрат і фермент (константа k–1):
Загальна концентрація ферменту в цьому випадку дорівнює |
|
[E] = [E0] + [ES]. |
(3.6) |
Швидкість зміни концентрації ферментFсубстратного комплексу складає
або з урахуванням (3.6)
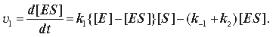 (3.7) Якщо в системі концентрація субстрату набагато перевищує концентрацію ферменту ([S]>>[E]), то кількість існуючих у кожний момент ферментFсубстратних комплексів залишається постійною (v1 =0), тобто система знаходиться в стаціонарному стані. У цьому випадку з (3.7)
(3.7) Якщо в системі концентрація субстрату набагато перевищує концентрацію ферменту ([S]>>[E]), то кількість існуючих у кожний момент ферментFсубстратних комплексів залишається постійною (v1 =0), тобто система знаходиться в стаціонарному стані. У цьому випадку з (3.7)
одержуємо концентрацію ферментFсубстратного комплексу
|
(3.8) |
де |
— константа Міхаеліса. |
|
З урахуванням (3.8) швидкість ферментативної реакції дорівнює |
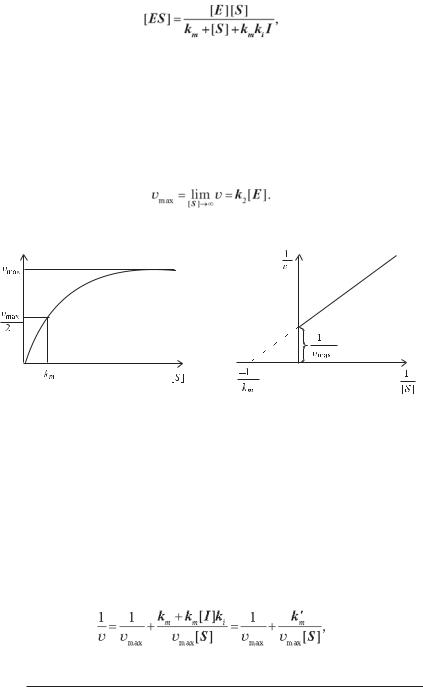
(3.9) Залежність швидкості ферментативної реакції від концентрації суб-
64

страту наведена на рисунку 3.20. При збільшенні концентрації субстрату
швидкість прагне до деякого максимального значення
Тоді вираз (3.9) прийме вигляд (рівняння Міхаеліса—Ментен)
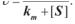 (3.10) Як бачимо, константа Міхаеліса чисельно дорівнює концентрації суб-
(3.10) Як бачимо, константа Міхаеліса чисельно дорівнює концентрації суб-
страту, при якій швидкість реакції дорівнює половині максимальної. Для кількісного визначення величин km і vmax рівняння (3.10) пере-
творюють до вигляду (метод Лайнуівера — Берка)
(3.11) Графік рівняння (3.11) наведений на рисунку 3.21. При 1/[S] = 0 одержуємо 1/v = 1/vmax, величина km визначається як тангенс кута на-
хилу прямої.
Деякі речовини, які зв’язуються із ферментом, зменшують швидкість ферментативної реакції (інгібітори) або збільшують (активатори). У якості інгібіторів або активаторів, що мають загальну назву модифікаторів, можуть виступати природні фізіологічні речовини, які регулюють ферментативну активність, а також цілий ряд лікарських препаратів.
Розрізняють конкурентні і неконкурентні інгібітори. Конкурентні інгібітори зв’язуються з активним центром ферменту, створюючи комплекс ферментFінгібітор ЕI, але в продукт не перетворюються.
У випадку присутності в системі конкурентного інгібітора, відповідно до схеми на рисунку 3.22,а, швидкість зміни концентрації комплексів ферментFсубстрат (ES) і ферментFінгібітор (EI) приймуть вигляд, відповідно
|
(3.12) |
|
(3.13) |
а загальна концентрація ферменту складе |
|
[E] = [E0] + [ES] + [EI]. |
(3.14) |
Вирішуючи спільно рівняння (3.12), (3.13) і (3.14) за умови встановлення стаціонарного стану, висловимо концентрацію ферментFсубстратного
65
комплексу в присутності конкурентного інгібітора
де ki = k3 /k–3.
Звідси швидкість ферментативної реакції в присутності конкурентного інгібітора складе
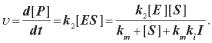 (3.15) У цьому випадку максимальна швидкість реакції не змінюється в порівнянні з максимальною швидкістю у відсутність конкурентного
(3.15) У цьому випадку максимальна швидкість реакції не змінюється в порівнянні з максимальною швидкістю у відсутність конкурентного
інгібітора
Однак при малих концентраціях субстрату швидкість реакції зменшується в порівнянні з тією, що була у відсутність конкурент-
Рис. 3.20. Залежність швидкості v
ферментативної реакції від концентрації Рис. 3.21. Графік Лайнуівера – Берка. субстрату [S].
ного інгібітора (рис. 3.23). Це можна пояснити тим, що при високих концентраціях субстрату ([S] →∞) фермент зв’язується переважно з ним, а не з інгібітором, концентрація якого виявляється значно нижче концентрації субстрату ([I] << [S]). При невисоких концентраціях субстрату [S]≈[I], фермент утворює комплекси як із субстратом, так і з інгібітором, що зменшує швидкість реакції. Для досягнення швидкості, що складає половину максимальної, концентрацію субстрату тепер потрібно збільшити в (1 + kiІ) раз.
У координатах Лайнуівера — Берка рівняння (3.15) прийме вигляд:
де k’m = km(1 + kiI) – константа Міхаеліса в присутності конкурентного
66

інгібітора (див. рис. 3.24, крива 2).
Неконкурентний інгібітор зв’язується із ферментFсубстратним комплексом, створюючи неактивний комплекс ESI. У цьому випадку,
а |
б |
Рис. 3.22. Схеми процесів із конкурентним (а) і неконкурентним (б) інгібіруванням.
відповідно до схеми на рисунку 3.22,б, швидкість зміни концентрації комплексів ферментFсубстрат (ES) і ферментFсубстратFінгібітор (ESI) складають, відповідно
а загальна концентрація ферменту
[E] = [E0] + [ES] + [ESI].
Тоді швидкість ферментативної реакції в стаціонарному стані в присутності неконкурентного інгібітора складе
де ki = k4 /k–4.
У цьому випадку максимальна швидкість реакції буде дорівнюва-
ти:
тобто зменшиться в порівнянні з максимальною швидкістю у відсутність неконкурентного інгібітора в (1 + kiІ) раз (рис. 3.24, крива 3).
Існує ряд ферментів, кінетика яких не підпорядковується рівнянню Міхаеліса — Ментен. Залежність швидкості ферментативної реакції від концентрації субстрату для них має вигляд, відмінної від гіперболи. У більшості випадків це явище можна пояснити таким чином.
Деякі ферменти складаються з кількох субодиниць і мають кілька центрів зв’язування субстрату. Приєднання субстрату до одного з центрів
67
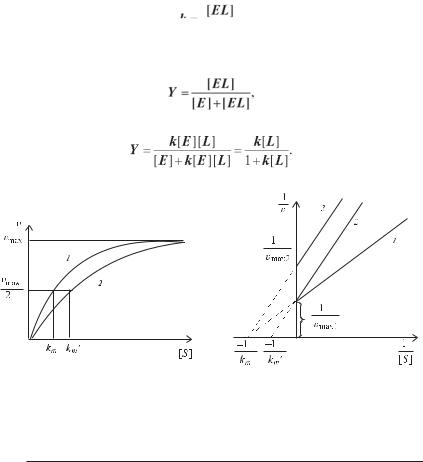
зв’язування викликає такі конформаційні перебудови у ферменті, що полегшують приєднання субстрату до наступного центру (так званий кооперативний ефект). У цьому випадку залежність швидкості ферментативної реакції від концентрації субстрату має SFобразний вигляд.
Розглянемо явище кооперативності на прикладі білка гемоглобіну, що складається з чотирьох субодиниць і чотирьох центрів зв’язування, і порівняємо його кінетику з міоглобіном, мономерним білком з одним центром зв’язування. Незважаючи на те, що ці білки являються не ферментами, а транспортними білками, що постачають організм киснем, кінетичні рівняння для них подібні до рівнянь для ферментів, що виявляють властивість кооперативності, як гемоглобін, або тих, що не виявляють, як міоглобін.
Реакцію утворення комплексу ліганду L із мономерним білком E можна представити як
де k — константа зв’язування, яка дорівнює
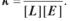 (3.16) Ступінь насичення білка лігандом, тобто відношення зайнятих
(3.16) Ступінь насичення білка лігандом, тобто відношення зайнятих
центрів зв’язування до їхнього загального числа, дорівнює:
або з урахуванням (3.16)
(3.17) |
Рис. 3.23. Залежність швидкості фермен- |
Рис. 3.24. Графік Лайнуівера – Берка: |
тативної реакції від концентрації суб- |
1 – без інгібітора; 2 – у присутності кон- |
страту у відсутність (1) і присутність (2) |
курентного інгібітора; 3 – у присутності |
конкурентного інгібітора. |
неконкурентного інгібітора. |
68

Рівняння (3.17) подібно рівнянню Міхаеліса — Ментен і графічно являє собою гіперболу (рис. 3.25, крива 1).
Для опису ступеня насичення гемоглобіну використовують рівняння, запропоноване Хіллом
(3.18)
де kh — константа зв’язування; h — параметр кооперативності.
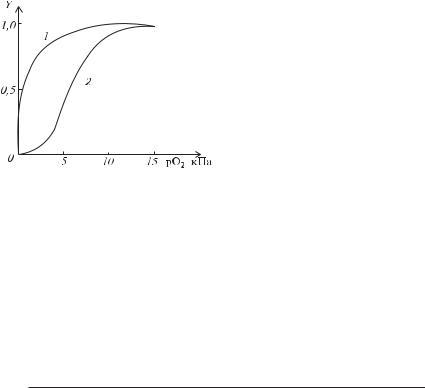
При h = 1 кооперативність відсутня і рівняння (3.18) приходить до вигляду (3.17). При h >1 кооперативність позитивна, тобто приєднання одного ліганду до центру зв’язування полегшує зв’язування з іншими, при h <1 кооперативність негативна. Для гемоглобіну h =2,8. На рисунку 3.25 наведена залежність насичення міоглобіну і гемоглобіну в залежності від парціального тиску кисню рО2, пропорційного концентрації.
У організмі парціальний тиск кисню змінюється в не дуже великих межах, однак кооперативні властивості гемоглобіну, що виявляються в SFподібній формі кривої приводять до того, що навіть при невеликих змінах парціального тиску кисню значно змінюється ступінь насичення гемоглобіну киснем. Якби гемоглобін не виявляв властивість кооперативності, то відщеплення кисню в тканинах відбувалося б не настільки інтенсивно.
§ 19. Біофізика нуклеїнових кислот
Будоваівластивостінуклеїновихкислотвизначаютьсяїхньоюфункцією
ворганізмі: збереженням і передачею генетичної інформації.
Уланцюзі дезоксирибонуклеїнової кислоти (ДНК) чергуються у певному порядку мономери — нуклеотиди, зв’язані між собою ковалентними фосфодіефірними зв’язками фосфатних груп із вуглеводами. Кожний із нуклеотидів має у своєму складі дезоксирибозу і залишок
фосфорної кислоти, і відрізняється від інших азотистою основою, яких у ДНК є 4 види: аденін (А ), гуанін (Г ), тимін (Т ) та цитозин (Ц ). Перші два з них є пуриновими, а другі — піримідиновими основами. Певна послідовність нуклеотидів у ланцюзі складає первинну структуру нуклеїнової кислоти.
Вторинна структура ДНК була розшифрована за допомогою рентгеноструктурного аналізу в 1952 р. Франклін, Кріком, Уотсоном та Уілкінсом. Молекула ДНК, як правило, складається з двох нуклеотидних
69
ланцюгів, лише в деяких вірусах зустрічаються одноланцюгові молекули ДНК. Два ланцюги ДНК зв’язані один з одним через азотисті основи водневими зв’язками, причому, аденін завжди утворює пару з тиміном, а гуанін — із цитозином (рис. 3.26). Говорять, що аденін комплементарний тиміну, а гуанін — цитозину, один ланцюг ДНК комплементарний іншому. Це пояснює правила Чаргафа, які були сформульовані раніше відкриття структури ДНК:
|
A +Г |
=1; |
|
А +Ц |
=1. |
1) А = Т; 2) Г = Ц; 3) T +Ц |
4) Г +Т |
||||
Крім водневих зв’язків між парами основ стабілізація структури ДНК досягається також міжплощинними взаємодіями основ (стекінгFвзаємодіями). Кожна комплементарна пара нуклеотидів повертається щодо попередньої на деякий кут навколо осі спіралі, в результаті утворюється вторинна структура ДНК — подвійна спіраль.
Модель Уотсона і Кріка пояснила явище самоподвоєння ДНК
— редуплікацію. У процесі редуплікації в ДНК розриваються водневі зв’язки між основами, і на кожному з двох ланцюгів будується новий, при цьому кожний материнський ланцюг використовується як матриця для дочірнього. Нові нуклеотиди приєднуються за принципом ком-
плементарності, тобто до аденіну приєднується тимін, до гуаніну
— цитозин, до тиміну — аденін, до цитозину — гуанін. Така модель редуплікації називається напівконсервативною, тому що кожна нова молекула ДНК містить у собі один материнський та
один дочірній ланцюг. Ланцюги ДНК можуть бути
Рис. 3.25. Криві насичення киснем міоглобіну лінійними або кільцевими. У
(1) і гемоглобіну (2) у залежності від пар- |
останніх — кінці молекул кова- |
|
ціального тиску кисню рО2. |
||
лентно замкнуті. Два нуклеотидні |
||
|
||
|
ланцюги однієї молекули ДНК |
завжди розташовуються антипаралельно: один ланцюг — від 3’F до 5’Fкінця, другий — навпаки (рис. 3.26).
Подвійна спіраль ДНК може існувати в різних конформаціях, перехід між якими здійснюється при зміні вологості кристалічних препаратів ДНК, солі ДНК та деяких інших параметрів, а також може бути викликаний взаємодією ДНК із білками й іншими компонентами клітини. Вважається, що фізіологічним умовам відповідає ВFформа ДНК. Характеристики АF, ВF і СFформ ДНК подані в таблиці 3.1, схематичні
70