

Общая характеристика и основные этапы создания антагонистов H2-рецепторов
Общая характеристика
Антисекреторные средства – лекарственные средства, уменьшающие выработку хлороводородной кислоты в париетальных клетках слизистой оболочки желудка.

К антисекреторным средствам относятся:
блокаторы H2-гистаминовых рецепторов,
ингибиторы протонной помпы,
М-холиноблокаторы,
производные простагландинов.
Антисекреторные средства применяют для лечения язвенной болезни желудка и 12-перстной кишки, гиперацидного гастрита, рефлюкс-эзофагита, синдрома Золлингера-Эллисона и других кислотозависимых заболеваний.
Антагонисты H2-гистаминовых рецепторов – группа антисекреторных лекарственных средств, уменьшающих выработку хлороводородной кислоты за счёт блокирования H2-рецепторов париетальных клеток слизистой оболочки желудка.
АТХ-классификация
A02B Противоязвенные лекарственные средства и лекарственные средства для лечения эзофагеального рефлюкса
A02BA Антагонисты H2-рецепторов
A02BA01 Циметидин
A02BA02 Ранитидин
A02BA03 Фамотидин
Основные этапы создания
1964 – Начало проекта поиска антигистаминных ЛС, уменьшающих выработку HCl в желудке (компания «Smith, Kline and French»).
Руководитель проекта – Sir James W. Black (1924-2010), создатель пропранолола, лауреат Нобелевской премии по физиологии и медицине 1988 г.
Открытие двух типов гистаминовых рецепторов
Установлено, что обычные антигистаминные лекарственные вещества, обладающие противоаллергическим действием, не уменьшают выработку HCl в желудке, следовательно, существует 2 типа гистаминовых рецепторов.
Введение в 5-е положение молекулы гистамина метильной группы увеличивает селективность по отношению к H2-рецепторам (электронодонорный заместитель в 5-м положении стабилизирует таутомер гистамина, имеющий более высокое сродство к таким рецепторам).
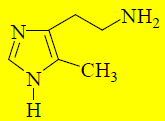
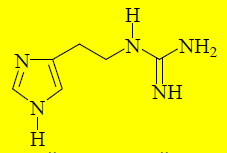
Производные гистамина, содержащие вместо –NH3+другие полярные группы будут связываться с тем же участком H2-рецептора, что и гистамин, и блокировать его. Гуанидиновый аналог гистамина проявляет свойства слабого антагониста по отношению к H2-рецепторам.
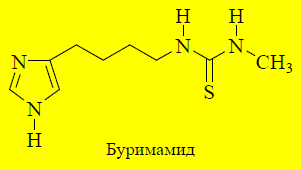
Увеличение длины боковой углеродной цепочки от двух до четырёх атомов и замена сильно основной гуанидиновой группы на нейтральный остаток тиомочевины заметно увеличивает способность соединения селективно блокировать H2-рецепторы. Такие соединения будут взаимодействовать только с сайтом антагонистов, но не агонистов. Синтезированный в 1972 году буримамид оказался достаточно высокоактивным антагонистом H2 -рецепторов, но из-за присутствия остатка тиомочевины обладал высокой токсичностью.
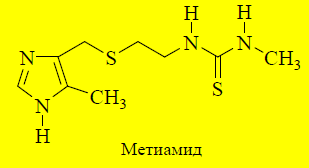
Уменьшение основности имидазольного ядра. Буримамид обладает более сильными основными свойствами ( pKBH+ = 7,25), чем гистамин (pKBH+ = 5,8). Введение электроноакцепторного заместителя в боковую цепь уменьшает основность. В 1972 году получено соединение, названное метиамидом(pKBH+ = 6,25). Антисекреторная активность данного вещества была в 10 раз выше, чем у буримамида.
Замена токсичного остатка тиомочевины на цианиминогруппуприводит к уменьшению токсичности. В 1973 году получен антагонист H2-рецепторов, названный циметидином (применяется в качестве лекарственного средства с 1976 года).
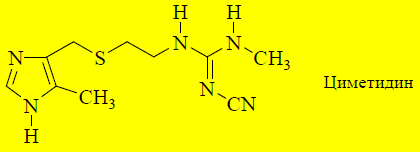

Циметидин оказался эффективным антисекреторным лекарственным средством, но имел и ряд существенных недостатков:
необходимость частого приёма,
антиандрогенная активность (может вызывать гинекомастию),
ингибирование цитохрома P-450 и увеличение активности других лекарственных веществ, которые подвергаются метаболическим превращениям с помощью данной группы ферментов.
Замена имидазольного ядра на биоизостеричные структуры
При дальнейшем поиске лекарственных веществ в ряду антагонистов H2-рецепторов установлено, что имидазольное кольцо не является обязательным элементом структуры таких соединений. Его замена на другой гетероцикл может привести к увеличению активности и селективности. Ранитидинсодержит в молекуле фурановый гетероцикл, во 2-м положении которого находится диметиламинометильная группа. Такое сочетание гетероцикла и боковой цепочки соответствует имидазольной структуре в молекуле циметидина. Ранитидин не обладает антиандрогенной активностью и лишь незначительно ингибирует цитохром Р-450.
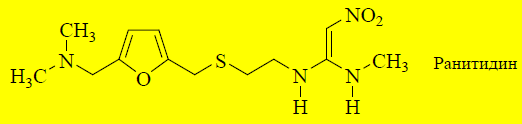
Ещё более эффективным антагонистом H2-рецепторов является фамотидин, в молекуле которого присутствует тиазольный цикл.
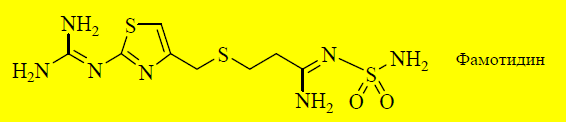
Фамотидин является более полярным соединением, чем циметидин и ранитидин, поэтому имеет меньшую биодоступность (40–45%). Он не обладает антиандрогенной активностью и не ингибирует цитохром Р-450.
Химическое строение и классификация антагонистов H2-рецепторов