

Лекция № 4. Кинетика химических реакций.
На прошлой лекции мы ввели критерий самопроизвольного протекания реакций (G<0), рассмотрели вопрос о принципиальной т.д. возможности или невозможности процесса. Однако, важно и то, насколько быстро будут протекать реакции.
Между значением G и скоростью нет зависимости (нельзя сказать, что чем больше по абсолютному значению G, тем быстрее достигается равновесие), так для реакций:
2NO (г) + O2(г) = 2NO2 (г); G0298= -70 кДж
2H2 (г) +O2(г)=2H2O(г); G0298 = -447 кДж
Первая реакция протекает очень быстро при комнатной температуре (образование бурого газа), а вторая реакция без катализатора практически не идет.
Если же с т.д. точки зрения реакция невозможна (G>0), то бессмысленно пытаться реализовать ее в данных условиях.
Исследованием течения реакций во времени занимается химическая кинетика. Она изучает скорости реакций, их зависимость от различных факторов и механизмы реакций.
Механизм реакции – последовательность и характер стадий хим. реакции.
По механизму различают простые и сложные реакции. Простые реакции осуществляются посредством простых элементарных актов. Элементарный акт – единичный акт взаимодействия частиц, в результате которого образуются новые частицы продуктов или промежуточных соединений. В элементарном акте принимает участие одна или две частицы, крайне редко – три.
Для сложных реакций необходимы разнотипные элементарные акты. Сложная реакция – суммарный результат нескольких элементарных процессов. Уравнение реакции в этом случае не отражает механизм реакции. Типы сложных реакций: параллельные (одновременно протекают два или несколько процессов), последовательные (А→В→С), сопряженные(А +В→ М, А+С→N), цепные.
Скорость химической реакции определяется изменением концентрации реагирующих веществ или продуктов реакции в единицу времени. (моль/л с)
Скорость реакции в общем случае не является постоянной в течение всего времени ее протекания. Скорость прямой реакции по мере расходования исходных веществ будет уменьшаться, скорость обратной – увеличиваться. Поэтому используют понятие средней скорости в данном интервале времени t:
vср = c/t
Истинная скорость (в любой момент времени) определяется первой производной концентрации по времени:
vср = dc/dt
(Рис. Фримантл, I, с. 414, кривая скорости)
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая

Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой (рис. 2.2); истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:

Скорость зависит:
-
от природы реагирующих веществ
-
от Т
-
От катализатора
-
От концентраций реагентов (для гомогенных)
-
От р (газы)
-
от площади соприкосновения
-
от среды (для реакций в растворах)
-
от формы сосуда (для цепных реакций)
-
от интенсивности света (для фотохимических реакций)
В 19 в. Гульдбергом и Вааге был сформулирован закон действующих масс –основной постулат хим. кинетики: скорость элементарной реакции пропорциональна произведению концентраций реагирующих веществ в степенях их стехиометрических коэффициентов.
v = k[A]a[B]b
Выражение зависимости скорости реакции от концентрации называют кинетическим уравнением.
Обратите внимание, что в формулировке закона присутствуют слова: для элементарной реакции. В приведенном виде закон работает только для элементарных реакций, т.е. для реакций, для которых стехиометрическое уравнение отражает механизм реакции. Пример: для реакции образования иодоводорода: v = k[I2][H2] (1967 показано, что эта реакция является сложной, не является бимолекулярной, реакция второго порядка. Как пример не годится).
Кинетическое уравнение в общем виде можно записать так:
v = k[A]p[B]q
p, q – порядки реакции по веществам А и В. (p+q)– суммарный порядок реакции.
В хим. кинетике реакции классифицируются по двум параметрам: по молекулярности и по порядку реакции.
Молекулярность – число молекул, участвующих в элементарном акте химического взаимодействия. Моно- би-, тримолекулярные. Вероятность одновременного соударения многих частиц мала, поэтому тримолекулярные реакции редки, а четырех – вообще неизвестны. (Молекулярность 3)
Для элементарных реакций порядок совпадает с молекулярностью. Для сложных реакций порядок не совпадает с молекулярностью.
Существуют реакции нулевого порядка (скорость не зависит от концентрации, например, разложение соединений на поверхности веществ, когда скорость не зависит от концентрации вещества в объеме). А также дробного порядка. ( медленные стадии имеют разный порядок, их скорости соизмеримы).
Порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени.
Необратимые реакции
Реакции нулевого порядка.
0-порядок по веществу встречается в гетерогенных реакциях. Для реакций нулевого порядка кинетическое уравнение имеет следующий вид:
![]() (II.5)
(II.5)
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения. [k]=моль/л с.

Реакции первого порядка.
А→В + С

(Рис. 2.6 Семиохин, с.23)
[k]=1/c
В качестве критерия скорости используют время полураспада – время, за которое концентрация реагента уменьшается в два раза. Для реакций первого порядка период полупревращения не зависит от концентрации исходного вещества.
Примеры необратимых реакций первого порядка.
2N2O5=4NO2 +O2
Это бимолекулярная реакция первого порядка( Карапетьянц, с. 233)
СH3OCH3 = CH4 + H2 + CO
Все реакции радиоактивного распада, гидролиз сложных эфиров в присутствии сильных кислот как катализаторов (это псевдопервый порядок за счет большого избытка воды)
Реакции второго порядка.
А + В → C + D


[k]=л/моль с
Реакции третьего порядка
А + В + С→ Е + F
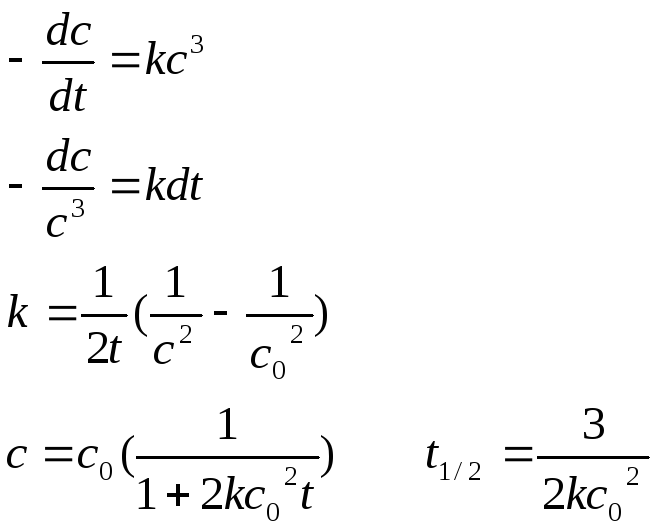
[k]=л2/моль2 с
Примеры: 2NO (г) + O2(г) = 2NO2 (г)
Дать графики со стр. 23 Семиохин.
Методы определения порядка реакции и констант скорости из экспериментальных данных.
Проведение реакции в условиях, когда концентрация одного из реагентов много меньше концентрации другого (других) и скорость реакции зависит от концентрации только этого реагента, используется для определения частных порядков реакции – это т.н. метод избыточных концентраций(избытка ) или метод изолирования Оствальда. Порядок реакции по данному веществу определяется одним из перечисленных ниже методов.
Графический метод (линеаризация) (lnc–t; 1/c –t; 1/c2–t)заключается в построении графика зависимости концентрации реагента от времени в различных координатах. Для различных частных порядков эти зависимости имеют следующий вид:
|
Порядок реакции |
Зависимость концентрации от времени |
|
1 |
|
|
2 |
|
|
3 |
|
Надо найти, в каких координатах график зависимости с=f(t) спрямляется. Если построить графики этих зависимостей на основании опытных данных, то лишь одна из них будет являться прямой линией. Если, например, график, построенный по опытным данным, оказался прямолинейным в координатах lnC = f(t), то частный порядок реакции по данному веществу равен единице.
Метод подстановки – подстановка данных в уравнения 1, 2 и 3 порядков, чтобы k=const. Метод подбора кинетического уравнения заключается в подстановке экспериментальных данных изучения зависимости концентрации вещества от времени в кинетические уравнения различных порядков. Подставляя в приведённые в таблице уравнения значения концентрации реагента в разные моменты времени, вычисляют значения константы скорости. Частный порядок реакции по данному веществу равен порядку того кинетического уравнения, для которого величина константы скорости остаётся постоянной во времени.
|
Порядок реакции |
Выражение для константы скорости |
|
1 |
|
|
2 |
|
|
3 |
|


Метод определения времени полупревращения заключается в определении t1/2 для нескольких начальных концентраций. Как видно из приведённых в таблице уравнений, для реакции первого порядка время полупревращения не зависит от Co, для реакции второго порядка – обратно пропорционально Co, и для реакции третьего порядка – обратно пропорционально квадрату начальной концентрации.
|
Порядок реакции |
Выражение для периода полупревращения |
|
1 |
|
|
2 |
|
|
3 |
|
По характеру зависимости t1/2 от Co нетрудно сделать вывод о порядке реакции по данному веществу. Дать рисунки с(t) из Фримантла. Данный метод, в отличие от описанных выше, применим и для определения дробных порядков.
Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Вант-Гоффом, сформулировавшим следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ. Математически правило Вант-Гоффа можно записать следующим образом:
![]() (II.29)
(II.29)
![]() (II.30)
(II.30)
Где =2-4.
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Уравнение Аррениуса описывает зависимость константы скорости реакции от температуры и величины энергии активации
![]() (II.36)
(II.36)
![]() (II.37)
(II.37)
Энергия активации рассчитывается из экспериментальных данных двумя способами: графическим и аналитическим. Надо построить график зависимости lnk от1/T. Тогда на оси абсцисс отсекается отрезок lnk0, а tg =-Ea/R.

Рис. 2.7 Зависимость логарифма константы скорости химической реакции от обратной температуры.
Аналитический способ предполагает сравнение скоростей при двух температурах:
lnk1=lnk0-Ea/RT1
lnk2=lnk0-Ea/RT2
lnk2/k1=Ea(T2-T1)/RT1T2
Механизмы реакций.
Последовательность отдельных стадий – механизм реакции.
О механизме реакции судят по экспериментальным данным, описывающим скорости протекания, а также по обнаружению промежуточных продуктов реакции.
Существует 2 подхода к рассмотрению элементарного акта.
-
Теория активных соударений
-
Теория активированного комплекса.
Теория активных соударений
Три постулата:
-
протекание реакции обусловлено столкновениями между реагирующими частицами
-
не каждое столкновение приводит к реакции, а только эффективное, столкновение между частицами, обладающими избытком энергии.
-
столкновение приводит к реакции только, если частицы определенным образом ориентированы в пространстве.
Энергия активации – та минимальная избыточная энергия теплового движения молекул в реакционной смеси, благодаря которой при столкновении возможно хим. превращение. Доля активных молекул в реакционной смеси обычно мала.(Рис из презентации МГУ или с.180, Коровин) На рис. показано распределение молекул по кинетической энергии – распределение Максвелла-Больцмана. Заштрихована доля молекул, обладающих энергией, выше Еа. При увеличении температуры растет доля молекул, обладающих избыточной энергией => увеличивается число столкновений.
В уравнении Аррениуса то, что стоит под exp – доля активных, благоприятных в энергетическом плане соударений. А – предэкспотенциальный множитель – P*Z. Z – число всех соударений. Р –стерический фактор, доля соударений, благоприятных в пространственном отношении. Р учитывает, что для взаимодействия сложных молекул необходима определенная ориентация. (этот метод не дает расчета энергии активации)
Теория активированного комплекса.
ТАК рассматривает превращение исходных веществ в продукты реакции через образование промежуточного активированного комплекса (переходного состояния). Она рассматривает систему реагирующих веществ с позиций квантовой механики. Подробно исследует геометрические изменения, происходящие в системе по мере образования продуктов из реагентов.
Рассмотрим путь некоторой элементарной реакции
А B + C ––> A…B…С→ А + ВС
Перераспределении связей происходит через образование активированного комплекса. Этот комплекс имеет избыток потенциальной энергии по сравнению с энергией реагирующих частиц. А затем энергия снижается по мере образования продуктов.
[Поскольку в реакции происходит разрыв одних связей и образование других, можно предположить, что энергия активации равна энергии разрыва хим. связи. Однако рез-ты измерений показывают, что она всегда меньше энергии связи. Для создания возможности протекания реакции нет необходимости в полном разрыве связей атомов в молекуле, надо их только несколько ослабить. Такое расшатывание связей происходит при образовании неустойчивого промежуточного соединения – активированного комплекса.
Разность между энергией активированного комплекса и средней энергией исходных молекул и есть энергия активации. Образование переходного состояния энергетически более выгодный процесс, чем полный распад вступающих в реакцию молекул (эн. активации меньше эн. диссоциации).]
Дать энтальпийные диаграммы – энергетические диаграммы реакции – с.237, Карапетьянц).

Рис. 2.5 Энергетическая диаграмма химической реакции. Eисх – средняя энергия частиц исходных веществ, Eпрод – средняя энергия частиц продуктов реакции
Сложные реакции
Сложные реакции протекают через несколько элементарных стадий: параллельные, последовательные, сопряженные, цепные.
-
Параллельные реакции – протекание нескольких процессов с участием одних и тех же исходных веществ. Эти процессы завершаются образованием разных продуктов реакции. Скорость определяется наиболее быстрой стадией.
KClO3 → KCl + O2
→ KClO4 + KCl
Наиболее быстрая реакция – главная, остальные – побочные.
-
Последовательные реакции – образование конечных продуктов происходит через ряд промежуточных продуктов. Скорость определяется самой медленной (лимитирующей стадией) (Рис презентация МГУ, с. 28)
2N2O5=4NO2 +O2
N2O5= NO2 +NO3 (быстрая)
NO2 + NO3 → NO2 +NO +O2 (медленная)
NO + NO3 → 2NO2 (быстрая)
v = kcN2O5 => реакция первого порядка
Cl2 +CHCl3 = HCl + CCl4
Cl2= 2Cl (быстрая)
Cl + CHCl3 → CCl3 + HCl (медленная)
Cl + CCl3 = CCl4 (быстрая)
v = kc1/2(Cl2)c(CHCl3)
Порядок реакции 1,5
-
Цепные реакции – реакции, которые вызываются свободными радикалами, под действием которых неактивные молекулы превращаются в активные.
Неразветвленные
Зарождение цепи:
Cl2 → 2Cl*
Развитие цепи
H2 + Сl* → HCl +H*
H* + Cl2 → HCl + Cl*
Обрыв цепи
H* + H* → H2
H* + Cl* → HCl
Обрыв на стенках сосуда.
Разветвленные – единичная реакция одного свободного радикала приводит к образованию больше, чем одного радикала.
H2 + O2 → 2HO*
H2 + HO* → H2O + H*
H*+O2 → HO* + O** (разветвление)
O* *+ H2 → H* + HO* (разветвление)
H* +HO* → H2O
Размножение радикалов приводит к лавинообразному течению процесса, может вызвать взрыв.
При нек-рых предельных (критических) условиях (р, Т) возможен внезапный переход от медленного течения процесса ко взрыву.
Хим. энцикл. т 5. Осн. понятие теории разветвленных Ц. р.- фактор разветвления цепи f, противоположный фактору гибели g переносчиков цепи в р-циях обрыва. Для всех этих р-ций характерно наличие нижнего и верхнего пределов самовоспламенения. Их проихождение определяется переходами через граничное условие. В случае газофазных р-ций при низком давлении диффузия переносчиков цепи к стенке и их гибель обуслоливает неравенство g >fи р-ция практически не наблюдается. Рост давления препятствует диффузии, при этом g уменьшается, а f растет, т.к. обычно в р-ции разветвления участвует один из реагентов, давление к-рого составляет часть общего давления. При переходе через граничное условие f = g происходит самоускорение р-ции и самовоспламенение смеси. Граничному условию соответствует нек-рое значение р1 – давление нижнего предела самовоспламенения. Соотношение f>g соблюдается при р>р1, но при дальнейшем росте р способность смеси к самовоспламенению исчезает. С ростом р растет доля тримолекулярных соударении, в результате к-рых происходит гибель переносчиков цепи в газовой фазе. Это м. б. также столкновения двух активных частиц с любой третьей частицей М и переход активной частицы в малоактивную, не участвующую в р-циях продолжения и разветвления цепи. Так, в смеси Н2 с О2 возможна гибель Н* по р-ции Н* + О2 + М —→ М + НО2* с послед, гибелью НО2* в р-циях друг с другом или с переносчиками цепи ОН* и Н*. За счет тримолекулярных р-ций фактор g, пропорциональный р2
опережает в своем росте фактор f, пропорциональный р. В результате при нек-ром р2 - давлении верхнего предела - вновь происходит переход через граничное условие f= g исмесь теряет способность к самовоспламенению. Явление верхнего предела было открыто и объяснено С. Хиншелвудом (1956). Семенову и Хиншелвуду за исследование механизма хим. р-ций была присуждена Нобелевская премия.
С ростом т-ры Т область воспламенения -- разность между р2 и р1 - расширяется, т.к. фактор f, характеризующий энергоемкую р-цию разветвления, возрастает с ростом Т значительно, а фактор g от Т зависит слабо. В случае понижения Т и р2 при нек-рой Т значения р1 и р2 становятся одинаковыми. Зависимости р1 ир2 от Т образуют характерный полуостров воспламенения (рис. 2). Контур этого
полуострова может изменяться при изменении условий опыта.
К цепным реакциям относится горение топлива. Для предотвращения взрыва в топливо вводят антидетонаторы – например, тетраэтилсвинец, взаимодействует с радикалами, обрывает цепь.
Катализ
Вещества, ускоряющие химическую реакцию – катализаторы.
Катализ – изменение скорости реакции под действием катализатора.
Ингибиторы – уменьшают скорость.
Катализатор вступает во взаимодействие с реагирующими веществами с образованием промежуточных продуктов и направляет процесс по новому реакционному пути. Механизм действия катализатора заключается в изменении пути процесса превращения. Новый путь характеризуется меньшей энергией активации.
1) Без кат:
А +В → А…В → АВ
2) С кат:
А + К АК
АК + В → В…АК
В…АК → АВ + К
(Рис. презентация МГУ, с 39)
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Действительно, если предположить, что предэкспоненциальные множители в уравнении Аррениуса (II.32) для каталитической и некаталитической реакций близки, то для отношения констант скорости можно записать:
 (II.44)
(II.44)
Катализатор не влияет на термодинамику, не изменяют энтальпию и энергию Гиббса, не влияет на константу равновесия.
Гомогенные каталитические реакции: все реагенты и катализатор в одной фазе.
Примеры:
СО +О2 → СО2 (кат – Н2О г)
СН3СНО → СН4 + СО (кат – I2 г)
Н2О2 → Н2О + О2 (тяжелые металлы, Fe2+, кровь, I-)
Гетерогенный катализ – реагирующая система и катализатор находятся в разных фазах.
Несколько стадий процесса
-
диффузия частиц к катализатору
-
адсорбция на поверхности катализатора
-
хим. реакция
-
десорбция
-
диффузия продуктов от поверхности
Н2 +О2 → Н2О (губчатая платина)
N2 +H2 → NH3 (Fe3O4, Cr2O3)
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Константа равновесия.
Обратимая хим. реакция – реакция, протекающая одновременно в противоположных направлениях.
Динамическое равновесие – скорость прямой = скорости обратной. После установления равновесия вещества имеют определенные равновесные концентрации. Условия равновесия – два противоположных процесса сбалансированы.
Для элементарных реакций можно вывести выражение для константы равновесия через кинетические представления.
Дать константы для р-ров и для газов.
Константа – мера глубины протекания процесса (степени превращения исходных веществ в конечные).
Константу равновесия можно выразить через термодинамические величины. Это более строгий подход, связывающий энергию Гиббса с К, позволяющий ответить на вопрос, будет ли реакция протекать самопроизвольно при данных концентрациях реагентов. Мы рассмотрим эти уравнения без вывода.
G0 = –RTlnKc
lnK = –G0 /RT
rG = rG0 +RTln(CcCCdD/CaACbB) – уравнение изотермы Вант-Гоффа
Влияние внешних условий на химическое равновесие
При постоянстве внешних условий система может находиться в состоянии равновесия сколь угодно долго. Если изменить эти условия (т.е. оказать на систему какое-либо внешнее воздействие), равновесие нарушается; в системе возникает самопроизвольный процесс, который продолжается до тех пор, пока система опять не достигнет состояния равновесия (уже при новых условиях). Рассмотрим, как влияют на положение равновесия некоторые факторы.
1.7.2 Влияние давления и концентрации
Рассмотрим несколько возможных случаев смещения равновесия.
1. В систему добавлено исходное вещество. В этом случае
![]() ;
; ![]() ;
;
По уравнению изотермы химической реакции (I.100 – I.101) получаем: ΔF < 0; ΔG < 0. В системе возникнет самопроизвольный химический процесс, направленный в сторону расходования исходных веществ и образования продуктов реакции (химическое равновесие смещается вправо).
2. В систему добавлен продукт реакции. В этом случае
![]() ;
;
![]() ;
;
Согласно уравнению изотермы химической реакции, ΔF > 0; ΔG > 0. Химическое равновесие будет смещено влево (в сторону расходования продуктов реакции и образования исходных веществ).
3. Изменено общее давление (для реакций в газовой фазе).
Парциальные давления всех компонентов Рi в этом случае изменяются в одинаковой степени; направление смещения равновесия будет определяться суммой стехиометрических коэффициентов Δn.
Учитывая, что парциальное давление газа в смеси равно общему давлению, умноженному на мольную долю компонента в смеси (Рi = РХi), изотерму реакции можно переписать в следующем виде (здесь Δn = Σ(ni)прод – Σ(ni)исх):
![]() (I.102)
(I.102)
![]() (I.103)
(I.103)
Примем, что Р2 > Р1. В этом случае, если Δn > 0 (реакция идет с увеличением числа молей газообразных веществ), то ΔG > 0; равновесие смещается влево. Если реакция идет с уменьшением числа молей газообразных веществ (Δn < 0), то ΔG < 0; равновесие смещается вправо. Иначе говоря, увеличение общего давления смещает равновесие в сторону процесса, идущего с уменьшением числа молей газообразных веществ. Уменьшение общего давления газов в смеси (Р2 < Р1) будет смещать равновесие в сторону реакции, идущей с увеличением числа молей газообразных веществ.
Необходимо отметить, что изменение концентрации или давления, смещая равновесие, не изменяет величину константы равновесия, которая зависит только от природы реагирующих веществ и температуры.
1.7.3 Влияние температуры на положение равновесия
Повышение либо понижение температуры означает приобретение либо потерю системой энергии и, следовательно, должно изменять величину константы равновесия.
Запишем уравнение (I.99) в следующем виде:
![]() (I.104)
(I.104)
![]() (I.105)
(I.105)
Продифференцировав выражение (I.105) по температуре, получаем для зависимости константы равновесия от температуры уравнение (I.106) – изобару Вант-Гоффа:
![]() (I.06)
(I.06)
![]()
Рассуждая аналогичным образом, для процесса, проходящего в изохорных условиях, можно получить изохору Вант-Гоффа:
![]() (I.107)
(I.107)
Изобара и изохора Вант-Гоффа связывают изменение константы химического равновесия с тепловым эффектом реакции в изобарных и изохорных условиях соответственно. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° < 0 (ΔU° < 0). В этом случае, согласно (I.106, I.107), температурный коэффициент логарифма константы равновесия отрицателен. Повышение температуры уменьшает величину константы равновесия, т.е. смещает равновесие влево.
Эндотермические реакции: ΔH° > 0 (ΔU° > 0). В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
С помощью указанных уравнений можно количественно оценить смещение хим. равновесия. Качественно оценить смещение равновесие можно с помощью принципа Ле-Шателье.
Действие рассмотренных нами факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия, называемый также принципом Ле Шателье:
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Направление протекания самопроизвольного процесса можно определить исходя из:
знака изменения энергии Гиббса
значения величины К (К>1 →, К<1 )
уравнения изотермы Вант-Гоффа
Из изобары Вант-Гоффа можно рассчитать
ΔH°, если известны К1, К2 и Т1, Т2 (узнать, экзо или эндотермическая реакция)
определить К1, если известны К2, Т1, Т2, ΔH°