

2.22. Метод Парризера-Попла-Парра
Объединение нулевого дифференциального перекрывания и -электронного приближения приводит к методу Парризера-Попла-Парра, дающего прекрасные результаты для -электронных систем. Матричные элементы оператора Фока для этого метода приведены в табл. 2.16. U равно потенциалу ионизации атома в соответствующем валентном состоянии, взятому с обратным знаком: - I . Одноцентровые кулоновские интегралы оцениваются по формуле Паризера-Парра (2.66). Величины I и A , необходимые для расчета U и определяют из спектроскопических данных для валентных состояний атомов, используя модель локализованных связей. Двухцентровые интегралы AB рассчитывают по формулам (2.67)-(2.68). Величины h считаются параметрами и выбираются по разному для расчета свойств основного (метод Попла) и возбужденных (метод Парризера-Парра) состояний.
Для основного состояния h являются резонансными интегралами = kS , где k подбирается так, чтобы наилучшим образом воспроизводить теплоты образования в представительном круге соединений (существует и ряд других параметризаций).
2) Для возбужденных состояний следует учесть, что волновая функция молекулы должна быть суммой волновых функций основного 0 и возбужденных i k состояний: = 0 + аi k i k. Обычно учитывают несколько однократно возбужденных электронных конфигураций заданной мультиплетности и ищут коэффициенты аi k вариационным методом. Матричные элементы ( 0 h 0) дают энергию основного состояния Е0, а элементы ( i k h j l) имеют вид:
( i k h j l) h i k, j l = kl k - ij i +2(jk li)-(jk il). (2.70)
В приближении НДП
h i k, j l = ij kl ( k - i) + [2(1-R)с j с l c i с k - с j с i с k c l], (2.71)
где R=0 для синглетных состояний и R=1 – для триплетных.
 Расчет
проводится методом ССП, причем вначале
определяют МО для основного состояния,
а затем, используя их, строят волновые
функции возбужденных состояний.
Расчет
проводится методом ССП, причем вначале
определяют МО для основного состояния,
а затем, используя их, строят волновые
функции возбужденных состояний.
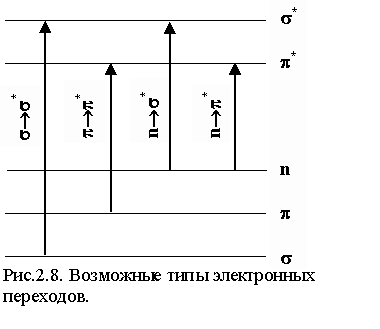
Метод Парризера-Попла-Парра включает в себя метод КВ и очень хорошо зарекомендовал себя как при определении геометрии, потенциалов ионизации и сродства к электрону, так и при расчетах оптических спектров поглощения сопряженных органических молекул. Спектр поглощения состоит из нескольких полос, связанных с определенными электронными переходами. Для плоских молекул МО можно разделить на три группы: , и n* . Наболее вероятное относительное расположение соответствующих энергетических уровней и разрешенные правилами отбора типы электронных переходов показаны на рис. 2.8. Диагонализизация матрицы h i k, j l называемой матрицей конфигурационного взаимодействия, дает энергии спектральных переходов и веса возбужденных конфигураций аi k. Ориентированный на -электроны, метод ППП хорошо описывает - * переходы (рис. 2.9.): точность оценки синглет-синглетных переходов составляет 0,1-0,2 эВ или 3-5% .
 Интенсивность
полосы поглощения f определяется
квадратом дипольного момента перехода
и равна f =К
аi
k c
i с
k
x
2.
Она оценивается методом ППП с ошибкой
40-50%.
Интенсивность
полосы поглощения f определяется
квадратом дипольного момента перехода
и равна f =К
аi
k c
i с
k
x
2.
Она оценивается методом ППП с ошибкой
40-50%.