Wypych Handbook of Solvents
.pdf5.5 The phenomenological theory of solvent effects |
301 |
5.5.4.4 Confounding effects
Solute-solute interactions
It is very commonly observed, in these mixed solvent systems, that the equilibrium solubility rises well above the dilute solution condition over some portion of the x2 range. Thus solution phase solute-solute interactions must make a contribution to G*soln . To some extent these may be eliminated in the subtraction according to eq. [5.5.22], but this operation cannot be relied upon to overcome this problem. Parameter estimates may therefore be contaminated by this effect. On the other hand, Khossravi25 has analyzed solubility data for biphenyl in methanol-water mixtures by applying eq. [5.5.23] over varying ranges of x2; he found that gA(γ2-γ1) was not markedly sensitive to the maximum value of x2 chosen to define the data set. In this system the solubility varies widely, from x3 = 7.1 x 10-7 (3.9 x 10-5 M) at x2 = 0 to x3 = 0.018 (0.43 M) at x2 = 1.
Coupling of general medium and solvation effects
In this theory the general medium and solvation effects are coupled through the solvation exchange constants K1 and K2, which determine the composition of the solvation shell surrounding the solute, and thereby influence the surface tension in the solvation shell. But the situation is actually more complicated than this, for if surface tension-composition data are fitted to eq. [5.5.26] the resulting equilibrium constants are not numerically the same as the solvation constants K1 and K2 evaluated from a solubility study in the same mixed solvent. Labeling the surface tension-derived constants K′1 and K′2 , it is usually found that K′1>K1 and K′2 >K2. The result is that a number attached to γ at some x2 value as a consequence of a nonlinear regression analysis according to eq. [5.5.23] will be determined by K1 and K2, and this number will be different from the actual value of surface tension, which is described by K′1 and K′2 . But of course the actual value of γ is driving the general medium effect, so the discrepancy will be absorbed into gA. The actual surface tension (controlled by K′1 and K′2 )
is smaller (except when x2 = 0 and x2 = 1) than that calculated with K1 and K2. Thus gapparent = gtrue x γ(K′1,K′2 )/γ(K1,K2). This effect will be superimposed on the curvature correction factor that g represents, as well as the direct coupling effect of solvation mentioned above.
The cavity surface area
In solubility studies of some substituted biphenyls, it was found (see 5.5.3.1) that gA evaluated via eq. [5.5.23] was linearly correlated with the nonpolar surface area of the solutes
rather than with their total surface area; the correlation equation was gA = 0.37 Anonpolar. It was concluded that the A in the parameter gA is the nonpolar surface area of the solute. This
conclusion, however, was based on the assumption that g is fixed. But the correlation equa-
tion can also be written gA = 0.37 FnonpolarAtotal, where Fnonpolar = Anonpolar/Atotal is the fraction of solute surface area that is nonpolar. Suppose it is admitted that g may depend upon the sol-
ute (more particularly, it may depend upon the solute’s polarity); then the correlation is con-
sistent with the identities A = Atotal and g = 0.37 Fnonpolar.
Thus differences in gA may arise from differences in solute polarity, acting through g. But A may itself change, rather obviously as a result of solute size, but also as a consequence of change of solvent, for the solvent size and geometry will affect the shape and size of the cavity that houses the solute.
302 |
Kenneth A. Connors |
The role of interfacial tension
In all the preceding discussion of terms having the gAγ form, γ has been interpreted as a surface tension, the factor g serving to correct for the molecular-scale curvature effect. But a surface tension is measured at the macroscopic air-liquid interface, and in the solution case we are actually interested in the tension at a molecular scale solute-solvent interface. This may be more closely related to an interfacial tension than to a surface tension. As a consequence, if we attempt to find (say) g2A2 by dividing g2A2γ2 by γ2, we may be dividing by the wrong number.
To estimate numbers approximating to interfacial tensions between a dissolved solute molecule and a solvent is conjectural, but some general observations may be helpful. Let γX and γY be surface tensions (vs. air) of pure solvents X and Y, and γXY the interfacial tension at the X-Y interface. Then in general,
γXY = γX + γY − WXY − WYX |
[5.5.56] |
where WXY is the energy of interaction (per unit area) of X acting on Y and WYX is the energy of Y acting on X. When dispersion forces alone are contributing to the interactions, this equation becomes26
γXY = γX + γY − 2(γXd γYd )1/ 2 |
[5.5.57] |
where γXd and γYd are the dispersion force components of γX and γY. In consequence, γXY is always smaller than the larger of the two surface tensions, and it may be smaller than either of them.
Referring now to Table 5.5.8, if we innocently convert g2A2γ2 values to estimates of g2A2 by dividing by γ2, we find a range in g2A2 from 23 Å2 molecule-1 (for dimethylsulfoxide) to 118 Å2 molecule-1 (for isopropanol). But if the preceding argument is correct, in dividing by γ2 we were dividing by the wrong value. Taking benzene (γ = 28 erg cm-2) as a model of supercooled liquid naphthalene, we might anticipate that those cosolvents in Table 5.5.8 whose γ2 values are greater than this number will have interfacial tensions smaller than γ2, hence should yield g2A2 estimates larger than those calculated with γ2, and vice versa. Thus, the considerable variability observed in g2A2 will be reduced.
On the basis of the preceding arguments it is recommended that gAγ terms (exemplified by g1A1γ1, g2A2γ2, and gA(γ2-γ1)) should not be factored into gA quantities through division by γ, the surface tension, (except perhaps to confirm that magnitudes are roughly as expected). This conclusion arises directly from the interfacial tension considerations.
Finally let us consider the possibility of negative gA values in eq. [5.5.23]. Eq. [5.5.54] shows that a negative gA is indeed a formal possibility, but how can it arise in practice? We take the water-ethanol-sucrose system as an example; gA was reported to be negative for this system. Water is solvent 1 and ethanol is solvent 2. This system is unusual because of the very high polarity of the solute. At the molecular level, the solute in contact with these solvents is reasonably regarded as supercooled liquid sucrose, whose surface tension is unknown, but might be modeled by that of glycerol (γ = 63.4 erg cm-2). In these very polar systems capable of hydrogen-bonding eq. [5.5.57] is not applicable, but we can anticipate that the sucrose-water interaction energies (the WXY and WXY terms in eq. [5.5.56] are
5.5 The phenomenological theory of solvent effects |
303 |
greater than sucrose-ethanol energies. We may expect that the sucrose-water interfacial tension is very low.
Now, gA turned out to be negative because gA(γ2-γ1), a positive quantity as generated by eq. [5.5.23], was divided by (γ2-γ1), a difference of surface tensions that is negative. Inevitably gA was found to be negative. The interfacial tension argument, however, leads to the conclusion that division should have been by the difference in interfacial tensions. We have seen that the interfacial tension between sucrose and water may be unusually low. Thus the factor (γ2-γ1), when replaced by a difference of interfacial tensions, namely [γ(sucrose/ethanol) - γ(sucrose/water)], is of uncertain magnitude and sign. We therefore do not know the sign of gA; we only know that the quantity we label gA(γ2-γ1) is positive. This real example demonstrates the soundness of the advice that products of the form gAγ not be separated into their factors.27,28
5.5.5 NOTES AND REFERENCES
1D. Khossravi and K.A. Connors, J. Pharm. Sci., 81, 371 (1992).
2 R.R. Pfeiffer, K.S. Yang, and M.A. Tucker, J. Pharm. Sci., 59, 1809 (1970).
3J.B. Bogardus, J. Pharm. Sci., 72, 837 (1983).
4P.L. Gould, J.R. Howard, and G.A. Oldershaw, Int. J. Pharm., 51, 195 (1989).
5 |
Also, when K1 = 1 and K2 = 1, eq. [5.5.13] shows that Gsolv = GWW; in this special case the solvation |
|
energy is composition-independent. |
6H.H. Uhlig, J. Phys. Chem., 41, 1215 (1937).
7 |
J.E. Leffler and E. Grunwald, Rates and Equilibria of Organic Reactions, J. Wiley & Sons, New York, |
|
1963, p. 22. |
8J.M. LePree, M.J. Mulski, and K.A. Connors, J. Chem. Soc., Perkin Trans. 2, 1491 (1994).
9The curvature correction factor g is dimensionless, as are the solvation constants K1 and K2. The parameter gA is expressed in Å2 molecule-1 by giving the surface tension the units J Å-2 (where 1 erg cm-2 = 1 x 10-23 J Å-2).
10D. Khossravi and K.A. Connors, J. Pharm. Sci., 82, 817 (1993).
11J.M. LePree, Ph.D. Dissertation, University of Wisconsin-Madison, 1995, p. 29.
12A. Leo, C. Hansch, and D. Elkins, Chem. Revs., 71, 525 (1971).
13C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, VCH, Weinheim, 1988.
14D. Khossravi and K.A. Connors, J. Solution Chem., 22, 321 (1993).
15K.A. Connors and J.L. Wright, Anal. Chem., 61, 194 (1989).
16K.A. Connors, Binding Constants, Wiley-Interscience, New York, 1987, pp. 51, 78.
17R.D. Skwierczynski and K.A. Connors, J. Chem. Soc., Perkin Trans. 2, 467 (1994).
18K.A. Connors and D. Khossravi, J. Solution Chem., 22, 677 (1993).
19M.J. Mulski and K.A. Connors, Supramol, Chem., 4, 271 (1995).
20K.A. Connors, M.J. Mulski, and A. Paulson, J. Org. Chem., 57, 1794 (1992).
21J.M. LePree and K.A. Connors, J. Pharm. Sci., 85, 560 (1996).
22M.C. Brown, J.M. LePree, and K.A. Connors, Int. J. Chem. Kinetics, 28, 791 (1996).
23J.M. LePree and M.E. Cancino, J. Chromatogr. A, 829, 41 (1998).
24The validity of this approximation can be assessed. The free energy of hydration of benzene is given as -0.77 kJ mol-1 (E. Grunwald, Thermodynamics of Molecular Species, Wiley-Interscience, New York, 1997,
p. 290). Doubling this to -1.5 kJ mol-1 because of the greater surface area of naphthalene and repeating the calculation gives g1A1γ1 = 4.88 x 10-20 J molecule-1, not sufficiently different from the value given in the text to change any conclusions.
25D. Khossravi, Ph.D. Dissertation, University of Wisconsin-Madison, 1992, p. 141.
26F.M. Fowkes, Chemistry and Physics of Interfaces; American Chemical Society: Washington, D.C., 1965, Chap. 1.
27The introduction of the interfacial tension into the cavity term was first done by Yalkowsky et al.,28 who also argue that a separate solute-solvent interaction term is unneeded, as the solute-solvent interaction is already embodied in the interfacial tension. In our theory we explicitly show the coupling between the solute-solvent and solvent-solvent interactions (eq. [5.5.19]), but this is in addition to the solute-solvent interaction (eq. [5.5.13]). This difference between the two theories is a subtle issue that requires clarification.
28S.H. Yalkowsky, G.L. Amidon, G. Zografi, and G.L. Flynn, J. Pharm. Sci., 64, 48 (1975).
6
Swelling
6.1MODERN VIEWS ON KINETICS OF SWELLING OF CROSSLINKED ELASTOMERS IN SOLVENTS
E. Ya. Denisyuk
Institute of Continuous Media Mechanics
V. V. Tereshatov
Institute of Technical Chemistry
Ural Branch of Russian Academy of Sciences, Perm, Russia
6.1.1 INTRODUCTION
Diffusion phenomena encountered in mass-transfer of low-molecular liquids play an important role in many technological processes of polymer manufacture, processing, and use of polymeric materials. Diffusion of organic solvents in crosslinked elastomers may cause considerable material swelling. In this case, the polymeric matrix experiences strains as large as several hundred percent, while a non-homogeneous distribution of a liquid due to diffusion results in establishing stress-strain state capable of affecting the diffusion kinetics. The processes of material deformation and liquid diffusion in such systems are interrelated and nonlinear in nature and are strongly dependent on physical and geometrical nonlinearities. Therefore, exact relations of nonlinear mechanics of elastic-deformable continuum are the mainstream of a sequential theory of mass-transfer processes of low-molecu- lar liquids in elastomers.
The general principles of the development of nonlinear models of mass transfer in elastically deformed materials were developed in studies.1,2 The general formulation of constitutive equations and the use of non-traditional thermodynamic parameters such as partial stress tensors and diffusion forces lead to significant difficulties in attempts to apply the theory to the description of specific objects.3,4 Probably, because of this, the theory is little used for the solution of applied problems.
In the paper,5 a theory for mechanical and diffusional processes in hyperelastic materials was formulated in terms of the global stress tensor and chemical potentials. The approach described in1,2 was used as the basic principle and was generalized to the case of a multi-component mixture. An important feature of the work5 is that, owing to the structure of constitutive equations, the general model can be used without difficulty to describe specific systems.
In the paper6 the nonlinear theory5 was applied to steady swelling processes of crosslinked elastomers in solvents. The analytical and numerical treatment reveals three

306 |
E. Ya. Denisyuk, V. V. Tereshatov |
possible mechano-diffusion modes which differ qualitatively. Self-similar solutions obtained for these modes describe asymptotic properties at the initial stage of swelling. These modes are related to thermodynamical material properties. The theoretical predictions have been verified in the experiments on real elastomers.
6.1.2 FORMULATION OF SWELLING FOR A PLANE ELASTOMER LAYER
Consider an infinite plane elastomer layer of thickness 2h embedded in a low-molecular liquid. Suppose that the elastomer initially does not contain liquid and is unstrained. This state is taken as a reference configuration. Let us introduce the Cartesian coordinates (x,y,z) with the origin placed in the layer center and relate them to a polymer matrix. In the examined problem, the Cartesian coordinates will be used as the material coordinates. With reference to the layer, the x axis has a transverse direction and the other axes have longitudinal directions. In our approach, we define the problem under consideration as a one-dimensional problem, in which all quantities characterizing the elastomer state depend only on the x-co- ordinate.
On swelling, the layer experiences transversal and longitudinal deformations which can be written as
X = X(x,t ) Y = ν(t )y Z = ν(t )z |
[6.1.1] |
where (X,Y,Z) are the spatial Cartesian coordinates specifying the actual configuration of the polymeric matrix. From this it follows that the relative longitudinal stretch of the layer is λ2 = λ3 = ν(t) and the relative transversal stretch is λ1 = λ(x,t) = ∂X / ∂x. The quantity
J = λ1λ2 λ3 = λν2 |
[6.1.2] |
characterizes a local relative change in the material volume due to liquid absorption.
The boundary conditions and the relations describing free swelling of the plane layer
in the reference configuration are represented in5 as |
|
|||||||||
|
∂N1 |
= |
∂ |
|
∂N1 |
|
N1 = N1 (x,t) |
|
||
|
|
|
|
D |
|
, |
[6.1.3] |
|||
|
∂t |
|
∂x |
|||||||
|
|
∂x |
|
|
|
|
||||
∂N2 / ∂t = 0 |
|
|
|
|
[6.1.4] |
|||||
|
∂σ1 / ∂x = 0 |
|
|
|
|
[6.1.5] |
||||
N1(x,0) = 0 |
|
|
|
|
[6.1.6] |
|||||
|
∂N1 (0,t) / ∂x = 0, |
X(0,t) = 0 |
[6.1.7] |
|||||||
µ(h,t) = 0, |
σ1 (h,t) = 0 |
[6.1.8] |
||||||||
|
σ2 (x,t) = σ3 (x,t) |
= 0 |
[6.1.9] |
|||||||
where: |
|
|
|
|
|
|
|
|
||
|
N1, N2 |
the molar concentrations of the liquid and the chains of polymeric network of elastomer, |
||||||||
µ |
|
|
|
respectively, |
|
|||||
|
|
|
the chemical potential of the liquid dissolved in material |
|
||||||
|
σk |
|
|
(k = 1,2,3) are the principal values of the Piola stress tensors. |
|
|||||

6.1 Modern views on kinetics of swelling |
307 |
The angular brackets denote integration with respect to coordinate x:
h
 K
K = h −1 ∫Kdx
= h −1 ∫Kdx
0
Owing to the symmetry of the swelling process in the layer, the problem is solved for 0<x< h.
The equation of the liquid transport [6.1.3] in a plane layer has the form of a general diffusion equation except for the diffusion coefficient of the liquid, which, in the general case, is defined by the function D = D(N1,ν), implying that it depends on the liquid concentration and the relative longitudinal stretch of the layer.5 Eq. [6.1.4] is the law of conservation of matter for the polymeric matrix, and Eq. [6.1.5] states that the process of elastomer swelling is in the state of mechanical equilibrium. The initial condition is explicitly defined by Eq. [6.1.6]. The constraint that the diffusion flux and the displacements of polymeric matrix along x-axis in the layer center are absent is given by Eq. [6.1.7]. Eq. [6.1.8] has the physical meaning that there exists a thermodynamical equilibrium at the elastomer-liquid interface and that elastomer is not subjected to transverse mechanical loading, while Eq. [6.1.9] means that the layer does not experience longitudinal stretch under the external force.
The assumption that the elastomer and the liquid are incompressible media can be mathematically represented by an incompressibility condition, which in the present case is written as5
J = φ−1 |
[6.1.10] |
where the volume fraction of the polymer is |
|
φ = N2V2 / (N1V1 + N2V2 ) |
[6.1.11] |
where:
V1 and V2 the molar volumes of liquid and chains of the elastomer network, respectively
To make the definition of the examined problem complete, we need to add to the above model equations, the constitutive relations for mechanical stress tensor and chemical potential of a liquid. According to5, 6 these equation are given by
σk = RTV2−1 (λk − I1λ−k1 / 3) − pJλ−k1 |
[6.1.12] |
µ = µ mix (φ) + RTZ −1φ1/ 3 Γ1 / 3 + V1p |
[6.1.13] |
µ mix = RT[ln(1− φ) + φ + χφ2 ] |
[6.1.14] |
where:
Rthe gas constant per mole
Tthe absolute temperature
µmix |
the chemical potential of mixing |
ppressure
χthe Flory-Huggins interaction parameter

308 |
E. Ya. Denisyuk, V. V. Tereshatov |
Z= V2/V1
I1 |
= λ21 + λ22 + λ23 |
Γ1 |
= I1/J2/3 |
The above equations follow from the classical high elasticity theory and the Flory theory of polymeric networks.7
From Eq. [6.1.5] and the second condition of Eq. [6.1.8], we find that σ1(x,t) = 0. This equation together with Eq. [6.1.12] yields the expression for pressure. By substituting it in the formulas for chemical potential [6.1.13] and longitudinal stresses, we find, using Eqs. [6.1.2] and [6.1.10], that
σ2 = σ3 = RTV2−1 (ν − J 2 / ν5 ) |
[6.1.15] |
µ = µ mix (1/ J) + RTZ −1J / ν4 |
[6.1.16] |
A substitution of Eq. [6.1.15] in Eq. [6.1.9] gives an expression for longitudinal stretch of the layer
ν6 = J 2 (x,t) |
[6.1.17] |
With consideration of Eq. [6.1.16], the boundary condition at x = h is transformed to
µ mix (1/ J)RT + Z −1J / ν4 = 0 |
[6.1.18] |
Thus, the initial swelling problem for a plane layer is reduced to a boundary value problem for diffusion equation [6.1.3] with boundary conditions of Eqs. [6.1.6], [6.1.7], [6.1.17] and [6.1.18]. The solution to this problem provides a full description of swelling processes in the plane layer. In other words, using Eqs. [6.1.1], [6.1.2], [6.1.10] and [6.1.15] we can define a current distribution of a liquid through the layer and calculate the stress-strain state of the material.
It should be noted that boundary conditions of Eq. [6.1.18] and Eq. [6.1.17] specify the existence of positive feedback in the system, which is responsible for the onset of unsteady boundary regime during material swelling. The nonlinear distributed systems with positive feedback are generally known as active media and are distinguished for their complex and multimode response.8 In free swelling, the response of elastomers is, in a sense, similar to that of active media. Such behavior is most pronounced when the extent of material swelling is high, which makes this case worthwhile for detailed investigation.
For high-swelling elastomers, the volume fraction of polymer in equilibrium swelling state denoted in the following as ε and the volume fraction of polymer at the elastomer-liq- uid interface φ= 1/J entering Eq. [6.1.18] are small quantities. The asymptotic behavior of the function µmix(φ) at ϕ → 0 is described by
µ mix (φ) / RT = −bφα |
[6.1.19] |
The constants b and α can be calculated using the Flory equation [6.1.14]. A second order expansion of ln(1-φ) as a power series of φgives b = 1/2-χ and α = 2. The scaling approach gives a slightly different value of α, which is found to be α = 9/4 (des Cloizeaux law9).

6.1 Modern views on kinetics of swelling |
309 |
A volume fraction of the polymer in equilibrium swelling state can be determined by substituting Eq. [6.1.19] in Eq. 6.1.18] and setting φ = J-1 = ε and ν = ε-1/3, yields ε ≈ (bZ )−3/ (3α−1) . Then, using Eqs. [6.1.18], [6.1.19] and the last relation, we arrive at the following expression for the volumetric swelling ratio of the layer at the elastomer-liquid interface:
J ≈ ε−1 (ε1/ 3ν)6d |
[6.1.20] |
||
where |
|
|
|
d = |
2 |
|
[6.1.21] |
|
|
||
3(α + 1) |
|||
Note that approximate Eq. 6.1.20] defines the strain dependence of the equilibrium swell ratio of the elastomer in a liquid medium under conditions of biaxial symmetric material extension.
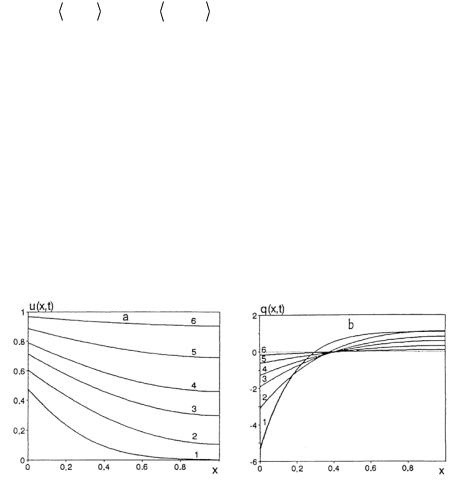
Substituting Eq. [6.1.17] in Eq. [6.1.20], we express the boundary swell ratio in terms of liquid distribution in the layer
J(h,t) = ε2 d −1 J 2 (x,t) d |
[6.1.22] |
|
Then the problem is finally defined as |
|
|
ut = (k(u,l)ux )x ; |
x (01,), t > 0 |
[6.1.23] |
u(x,0) = 0, ux (1,t) = 0 |
[6.1.24] |
|
(1− ε)u(0,t) + ε = |
[(1− ε)u(x,t) + ε]2 d |
[6.1.25] |
Here we assign dimensions to the variables. The quantities h and h2/D0 (where D0 is the value of diffusion coefficient in the state of ultimate elastomer swelling) are used as the units of distance and time. For the sake of convenience we transform, the coordinate to x→1-x. Integrating for x between the limits from 0 to 1 in Eq. [6.1.25] is designated by angular brackets. The function u(x,t) takes the value over the interval (0,1) and represents a dimensionless concentration of penetrating liquid. It is related to the liquid concentration and local material swelling by the following equations:
N1 =V1(ε−1 − 1)u(x,t), J(x,t) = ε−1 [(1− ε)u(x,t) + ε] |
[6.1.26] |
The quantity l = ε1/ 3ν represents the longitudinal layer stretch normalized to unity. By virtue of [6.1.17] and [6.1.26] we may write
l 6 (t) = [(1− ε)u(x,t) + ε]2 |
[6.1.27] |