

Wasserscheid P., Welton T. - Ionic Liquids in Synthesis (2002)(en)
.pdf256 |
Peter Wasserscheid |
|
|
|
|
|
J. Howarth, Tetrahedron Lett. 2000, |
|
1998 [Chem. Abstr. 1998, 129, 149094]. |
59 |
|
|||
|
|
41(34), 6627–6629. |
76 |
P. Wasserscheid, H. Waffenschmidt, |
60 |
S. V. Ley, C. Ramarao, M. D. Smith, |
|
P. Machnitzki, K. Kottsieper, O. Stel- |
|
|
|
Chem. Commun. 2001, 2278–2279. |
|
zer, Chem. Commun. 2001, 451–452. |
61 |
G. S. Owens, M. M. Abu-Omar, Chem. |
77 |
C. P. Casey, G. T. Whiteker, M. G. |
|
|
|
Commun. 2000, 1165–1166. |
|
Melville, L. M. Petrovich, L. J. A. |
62 |
C. E. Song, E. J. Roh, Chem. Commun. |
|
Gavey, D. R. J. Powell, J. Am. Chem. |
|
|
|
2000, 837–838. |
|
Soc. 1992, 114, 5535–5543. |
63 |
L. Gaillon, F. Bedioui, Chem. |
78 |
M. Kranenburg, Y. E. M. van der |
|
|
|
Commun. 2001, 1458–1459. |
|
Burgt, P. C. J. Kamer, P. W. N. M |
64 |
R. D. Singer, P. J. Scammells, Tetra- |
|
van Leeuwen, K. Goubitz, J. Fraanje, |
|
|
|
hedron Lett. 2001, 42(39), 6831–6833. |
|
Organometallics 1995, 14, 3081–3089. |
65 |
a) E. G. Kuntz, E. Kuntz, DE 2627354 |
79 |
P. W. N. M. van Leeuwen, P. C. J. |
|
|
|
(to Rhone-Poulenc S. A., Fr.), 1976, |
|
Kamer, J. N. H. Reek, P. Dierkes, |
|
|
[Chem. Abstr. 1977, 87, 101944]; |
|
Chem. Rev. 2000, 100, 2741–2770. |
|
|
b) E. G. Kuntz, CHEMTECH 1987, 17, |
80 |
P. Dierkes, S. Ramdeehul, L. Barloy, |
|
|
570–575; c) B. Cornils, W. A. Herr- |
|
A. De Cian, J. Fischer, P. C. J. Kamer, |
|
|
mann, “Aqueous-Phase Organo- |
|
P. W. N. M. van Leeuwen, Angew. |
|
|
metallic Catalysis“, Wiley-VCH, |
|
Chem., Int. Ed. 1998, 37, 3116–3118. |
|
|
Weinheim, 1998. |
81 |
F. Favre, H. Olivier-Bourbigou, |
66 |
P. Wasserscheid, H. Waffenschmidt, |
|
D. Commereuc, L. Saussine, Chem. |
|
|
|
J. Mol. Catal. A: Chem. 2000, 164(1–2), |
|
Commun. 2001, 1360–1361. |
|
|
61–67. |
82 |
J. Sirieix, M. Ossberger, B. Betze- |
67 |
Y. Chauvin, H. Olivier, L. Mußmann, |
|
meier, P. Knochel, Synletters 2000, |
|
|
|
Y. Chauvin, EP 776880 (to Institut |
|
1613–1615. |
|
|
Français du Pétrole, Fr.), 1997 [Chem. |
83 |
a) K. W. Kottsieper, O. Stelzer, |
|
|
Abstr. 1997, 127, 65507]. |
|
P. Wasserscheid, J. Mol. Catal. A: |
68 |
C. C. Brasse, U. Englert, A. Salzer, |
|
Chem. 2001, 175(1–2), 285–288; |
|
|
|
H. Waffenschmidt, P. Wasserscheid, |
|
b) D. J. Brauer, K. W. Kottsieper, |
|
|
Organometallics 2000, 19(19), |
|
C. Liek, O. Stelzer, H. Waffenschmidt, |
|
|
3818–3823. |
|
P. Wasserscheid, J. Organomet. Chem. |
69 |
J. D. Unruh, R. Christenson, J. Mol. |
|
2001, 630(2), 177–184. |
|
|
|
Catal. 1982, 14, 19–34. |
84 |
M. F. Sellin, P. B. Webb, D. J. Cole- |
70 |
C. P. Casey, E. L. Paulsen, E. W. Beut- |
|
Hamilton, Chem. Commun. 2001, |
|
|
|
tenmueller, B. R. Proft, L. M. Petro- |
|
781–782. |
|
|
vich, B. A. Matter, D. R. Powell, J. Am. |
85 |
D. E. Kaufmann, M. Nouroozian, |
|
|
Chem. Soc. 1997, 119, 11817–11825. |
|
H. Henze, Synlett 1996, 1091–1092. |
71 |
W. R. Moser, C. J. Papile, D. A. Bran- |
86 |
a) W. A. Herrmann, V. P. W. Böhm, |
|
|
|
non, R. A. Duwell, J. Mol. Catal. 1987, |
|
J. Organomet. Chem. 1999, 572, |
|
|
41, 271–292. |
|
141–145; b) V. P. W. Böhm, W. A. |
72 |
A. Heßler, O. Stelzer, H. Dibowski, |
|
Hermann, Chem. Eur. J. 2000, 6, |
|
|
|
K. Worm, F. P. Schmidtchen, J. Org. |
|
1017–1025. |
|
|
Chem. 1997, 62, 2362–2369. |
87 |
S. Bouquillon, B. Ganchegui, B. |
73 |
P. Machnitzki, M. Teppner, K. Wenz, |
|
Estrine, F. Henin, J. Muzart, J. Orga- |
|
|
|
O. Stelzer, E. J. Herdtweck, |
|
nomet. Chem. 2001, 634, 153–156. |
|
|
Organomet. Chem. 2000, 602, 158–169. |
88 |
a) V. Calò, A. Nacci, L. Lopez, A. |
74 |
H. Dibowski, F. P. Schmidtchen, |
|
Napola, Tetrahedron Lett. 2001, 42, |
|
|
|
Angew. Chem., Int. Ed. 1998, 37, |
|
4701–4703; b) V. Calo, A. Nacci, |
|
|
476–478. |
|
A. Monopoli, L. Lopez, A. di Cosmo, |
75 |
O. Stelzer, F. P. Schmidtchen, A. |
|
Tetrahedron, 2001, 57, 6071–6077. |
|
|
|
Heßler, M. Tepper, H. Dibowski, H. |
89 |
J. Silberg, T. Schareina, R. Kempe, |
|
|
Bahrmann, M. Riedel, DE 19701245 |
|
K. Wurst, M. R. Buchmeiser, J. Orga- |
|
|
(to Celanese G.m.b.H., Germany), |
|
nomet. Chem. 2001, 622, 6–18. |
|
|
5.2 Transition Metal Catalysis in Ionic Liquids |
257 |
|
|
|
|
|
|
90 |
L. Xu, W. Chen, J. Ross, J. Xiao, Org. |
103 |
W. Keim, B. Hoffmann, R. Lodewick, |
|
|
Lett. 2001, 3(2), 295–297. |
|
M. Peukert, G. Schmitt, J. Fleisch- |
|
91 |
A. J. Carmichael, M. J. Earle, J. D. |
|
hauer, U. Meier, J. Mol. Catal. 1979, |
|
|
Holbrey, P. B. McCormac, K. R. Sed- |
|
6, 79–97. |
|
|
don, Org. Lett. 1999, 1, 997–1000. |
104 |
a) B. Ellis, W. Keim, P. Wasserscheid, |
|
92 |
R. R. Deshmukh, R. Rajagopal, K. V. |
|
Chem. Commun. 1999, 337–338; |
|
|
Srinivasan, Chem. Comm. 2001, |
|
b) P. Wasserscheid, W. Keim, |
|
|
1544–1545. |
|
WO 9847616 (to BP Chemicals), 1997 |
|
93 |
H. Hagiwara, Y. Shimizu, T. Hoshi, |
|
[Chem. Abstr. 1998, 129, 332457]. |
|
|
T. Suzuki, M. Ando, K. Ohkubo, |
105 |
a) M. Eichmann, dissertation, RWTH- |
|
|
C. Yokoyama, Tetrahedron Lett. 2001, |
|
Aachen, 1999; b) P. Wasserscheid, |
|
|
42(26), 4349–4351. |
|
M. Eichmann, Catal. Today 2001, |
|
94 |
C. J. Mathews, P. J. Smith, T. Welton, |
|
66(2–4), 309–316. |
|
|
Chem. Comm. 2000, 1249–1250. |
106 |
a) I. Brassat, Ph.D. thesis, RWTH |
|
95 |
S. T. Handy, X. Zhang, Org. Lett. 2001, |
|
Aachen, 1998; b) I. Brassat, W. Keim, |
|
|
3(2), 233–236. |
|
S. Killat, M. Möthrath, P. Mastrorilli, |
|
96 |
G. Wilke, B. Bogdanovic, P. Hardt, |
|
C. Nobile, G. J. Suranna, Mol. Catal. |
|
|
P. Heimbach, W. Keim, M. Kröner, |
|
A: Chem. 2000, 43, 41–58; I. Brassat, |
|
|
W. Oberkirch, K. Tanaka, E. Stein- |
|
U. Englert, W. Keim, D. P. Keitel, S. |
|
|
rücke, D. Walter, H. Zimmermann, |
|
Killat, G. P. Suranna, R. Wang, Inorg. |
|
|
Angew. Chem. 1966, 5, 151–154. |
|
Chim. Acta 1998, 280, 150–162. |
|
97 |
a) Y. Chauvin, B. Gilbert, I. Guibard, |
107 |
P. Wasserscheid, C. Hilgers, submit- |
|
|
J. Chem. Soc. Chem. Commun. 1990, |
|
ted for publication. |
|
|
1715–1716; b) Y. Chauvin, S. Einloft, |
108 |
S. M. Silva, P. A. Z. Suarez, R. F. de |
|
|
H. Olivier, Ind. Eng. Chem. Res. 1995, |
|
Souza, J. Dupont, Polymer Bull. 1998, |
|
|
34, 1149–1155; c) Y. Chauvin, S. |
|
41, 401–405. |
|
|
Einloft, H. Olivier, FR 93/11,381 (to |
109 |
R. A. Ligabue, R. F. de Souza, |
|
|
Institut Français du Pétrole, Fr.), 1996 |
|
J. Dupont, J. Mol. Catal. A: Chem. |
|
|
[Chem. Abstr. 1995, 123, 144896c]. |
|
2001, 169, 11–17. |
|
98 |
a) M. Freemantle, Chem. Eng. News |
110 |
a) C. Gurtler, M. Jautelat, EP 1035093 |
|
|
1998, 76(13), 32–37; b) E. Burridge, |
|
(to Bayer A.G., Germany), 2000 |
|
|
ECN Chemscope 1999, May, 27–28; |
|
[Chem. Abstr. 2000, 133, 237853]; |
|
|
c) H. Olivier, J. Mol. Catal. A: Chem. |
|
b) R. C. Buijsman, E. van Vuuren, |
|
|
1999, 146(1–2), 285–289. |
|
J. G. Sterrenburg, Org. Lett. 2001, |
|
99 |
S. Einloft, F. K. Dietrich, R. F. de |
|
3(23), 3785–3787; c) D. Semeril, H. |
|
|
Souza, J. Dupont, Polyhedron 1996, |
|
Olivier-Bourbigou, C. Bruneau, P. H. |
|
|
19, 3257–3259. |
|
Dixneuf, Chem. Comm. 2002, 146–147. |
|
100 |
H. Olivier, P. Laurent-Gérot, J. Mol. |
111 |
E. Mizushima, T. Hayashi, M. Tanaka, |
|
|
Catal. A: Chem. 1999, 148, 43–48. |
|
Green Chem. 2001, 2, 76–79. |
|
101 |
J. T. Dixon, J. J. C. Grove, A. Ranwell, |
112 |
J. Ross, W. Chen, L. Xu, J. Xiao, |
|
|
WO 0138270 (to Sasol Technology |
|
Organometallics 2001, 20, 138–142. |
|
|
(Pty) Ltd, S. Afr.), 2001 [Chem. Abstr. |
113 |
S. Toma, B. Gotov, I. Kmentova, |
|
|
2001, 135, 7150]. |
|
E. Solcaniova, Green Chem. 2000, 2, |
|
102 |
a) Y. Chauvin, H. Olivier, C. N. Wyr- |
|
149–151. |
|
|
valski, L. C. Simon, R. F. de Souza, |
114 |
a) W. Chen, L. Xu, C. Chatterton, |
|
|
J. Catal. 1997, 165, 275–278; b) L. C. |
|
J. Xiao, Chem. Commu., 1999, |
|
|
Simon, J. Dupont, R. F. de Souza, |
|
1247–1248; b) C. de Bellefon, E. Pollet, |
|
|
J. Mol. Catal. 1998, 175, 215–220. |
|
P. Grenouillet, J. Mol. Catal. 1999, |
|
|
|
|
145, 121–126. |
|

258Hélène Olivier-Bourbigou, Alain Forestière
5.3
Ionic Liquids in Multiphasic Reactions
Hélène Olivier-Bourbigou and Alain Forestière
5.3.1
Multiphasic Reactions: General Features, Scope, and Limitations
While the solubility of organometallic complexes in common organic solvents appears to be an advantage in terms of site availability and tunability, reaction selectivity, and activity, it is a major drawback in terms of catalyst separation and recycling. The quest for new catalyst immobilization or recovery strategies to facilitate reuse is unceasing. Immobilization of the catalyst on a solid support has been widely studied. Except for Ziegler–Natta- and metallocene-type polymerization processes, in which the catalyst is not recycled due to its high activity, this technology has not yet been developed industrially, mainly because of problems of catalyst leaching and deactivation. One successful approach to close the advantage/disadvantage gap between homogeneous and heterogeneous catalysis is multiphasic catalysis [1]. In its simplest version, there are only two liquid phases (“biphasic” catalysis or “twophase” catalysis). The catalyst is dissolved in one phase (generally a polar phase), while the products and the substrates are found in the other. The catalyst can be separated by decantation and recycled under mild conditions.
It is important to make the distinction between the multiphasic catalysis concept
and transfer-assisted organometallic reactions or phase-transfer catalysis (PTC). In this latter approach, a catalytic amount of quaternary ammonium salt [Q]+[X]– is present in an aqueous phase. The catalyst’s lipophilic cation [Q]+ transports the reactant’s anion [Y]– to the organic phase, as an ion-pair, and the chemical reaction occurs in the organic phase of the two-phase organic/aqueous mixture [2].
The use of multiphasic catalysis has proven its potential in important industrial processes. In 1977, the first large-scale commercial catalytic process to benefit from
two-phase liquid/liquid technology was the Shell Higher Olefin Process (SHOP) for oligomerization of ethene into α-olefins, catalyzed by nickel complexes dissolved in diols such as 1,4-butanediol. Subsequently, the advancement in two-phase homogeneous catalysis has been demonstrated by the introduction of biphasic aqueous hydroformylation as an economically competitive large-scale process. The first commercial oxo plant, developed by Ruhrchemie–Rhône–Poulenc for the production of butyraldehyde from propene, came on stream in 1984. This is an example of a gas- liquid-liquid multiphasic system in which the homogeneous rhodium-based catalyst is immobilized in a water phase thanks to its coordination to the hydrophilic trisulfonated triphenylphosphine ligand (TPPTS) [3]. The catalyst separation is more effective and simpler than in classical rhodium processes, but separation of by-products from the catalyst is also an important issue.
Since then, water has emerged as a useful solvent for organometallic catalysis. In addition to the hydroformylation reactions, several other industrial processes
5.3 Ionic Liquids in Multiphasic Reactions |
259 |
|
|
employing homogeneous catalysis have been converted to aqueous-phase procedures [4].
5.3.2
Multiphasic Catalysis: Limitations and Challenges
Multiphasic (biphasic) catalysis relies on the transfer of organic substrates into the catalyst phase or on catalysis at the phase boundary. Most organic substrates do not have sufficient solubility in the catalyst phase (particularly in water) to give practical reaction rates in catalytic applications. Therefore, although the use of aqueousbiphasic catalysis has proven its potential in important industrial processes, the current applications of this technique remain limited: firstly to catalysts that are stable in the presence of water, and secondly to substrates that have significant water solubility. Many studies have focused on improving the affinities between the two liquid aqueous/organic phases, either through increasing the lipophilic character of the catalyst phase or even by immobilizing the catalyst on a support. For example, rapid stirring, emulsification, and sonication have all been used to increase the interfacial area. The addition of co-solvents to the aqueous phase has been investigated extensively as a means to improving the solubility of higher olefinic substrates in the catalyst-containing phase. Application of detergents or micellar processes to promote substrate transfer to the interface, or the addition of co-lig- ands such as PPh3 – or even of ligands with an amphiphilic character or modified cyclodextrins – also play rate-enhancing roles. The development of supported aque- ous-phase catalysis (SAPC), which involves the dissolution of an aqueous-phase complex in a thin layer of water adhering to a silica surface, opens the way to the reactivity of hydrophobic substrates. Although all these techniques can change the solubility of organic substrates in the aqueous phase or favor the concentration of the active center at the interface, they can also cause the leaching of a proportion of the catalyst into the organic phase.
The major advantage of the use of two-phase catalysis is the easy separation of the catalyst and product phases. However, the co-miscibility of the product and catalyst phases can be problematic. An example is given by the biphasic aqueous hydroformylation of ethene to propanal. Firstly, the propanal formed contains water, which has to be removed by distillation, This is difficult, due to formation of azeotropic mixtures. Secondly, a significant proportion of the rhodium catalyst is extracted from the reactor with the products, which prevents its efficient recovery. Nevertheless, the reaction of ethene itself in the water-based Rh-TPPTS system is fast. It is the high solubility of water in the propanal that prevents the application of the aqueous biphasic process [5].
To overcome these limitations, there has been a great deal of investigation of novel methods, one of them focused on the search for alternative solvents [6, 7]. Table 5.3-1 gives different approaches to biphasic catalysis, with some of their respective advantages and limitations.
Although already well known, perfluorinated solvents have only quite recently proved their utility in many organic and catalyzed reactions. The main advantage of

260Hélène Olivier-Bourbigou, Alain Forestière
Table 5.3-1: Advantages and limitations of different approaches for multiphasic “homogeneous” catalysis.
Catalyst phase |
Product phase |
Advantages |
Limitations |
||
|
|
|
|
|
|
Water |
Organic liquid |
■ |
Easy product separation |
■ |
Low reaction rate for |
(+co-solvent) |
|
|
and catalyst recycling |
|
poorly water-miscible |
|
|
■ |
Lower cost of chemical |
|
substrate |
|
|
|
processes |
■ |
Mass transfer limits |
|
|
■ Lack of toxicity of water |
|
rate of reaction |
|
|
|
|
|
■ |
Treatment of spent water |
Polar solvent |
Organic liquid |
■ |
Solvent effect |
■ |
Use of volatile organic |
|
|
|
|
|
solvent |
|
|
|
|
■ |
Co-miscibility of the two |
|
|
|
|
|
phases |
Fluorinated |
Organic liquid |
■ |
Temperature dependency |
■ |
Solvent and ligand |
organic solvent |
|
|
of the miscibility of |
|
costs |
|
|
|
fluorinated phase with |
■ |
Product contamination |
|
|
|
organic solvents |
|
|
Water |
Supercritical |
■ |
Organic co-solvent |
■ |
Poor solvating ability |
|
fluids (e.g. CO2) |
|
not needed |
|
of supercritical fluids |
|
|
■ |
High miscibility of |
■ |
High investment and |
|
|
|
CO2 with gas |
|
operating costs |
Ionic liquid |
Organic liquid |
■ |
Tunability of the solubility |
■ |
Ionic liquid costs |
|
|
|
characteristics of the |
■ |
Disposal of spent |
|
|
|
ionic liquids |
|
ionic liquids |
|
|
■ |
Solvent effect |
|
|
Ionic liquid |
Supercritical |
■ |
Organic co-solvent not |
■ |
Ionic liquid costs |
|
fluids (e.g. CO2) |
|
needed |
■ |
High pressure apparatus |
|
|
■ Tunability of the solubility |
|
needed |
|
|
|
|
characteristics of the |
|
|
|
|
|
ionic liquids |
|
|
|
|
■ |
Presence of CO2 reduces |
|
|
|
|
|
ionic liquid’s viscosity |
|
|
|
|
|
|
|
|
these solvents is that their miscibility with organic products can be tuned by variation of the temperature. Fluorous-phase catalysis makes possible the association of homogeneous phase catalysis (thus avoiding problems of mass-transfer limitations) and a biphasic separation of the catalyst and reaction mixture [8]. However, these solvents are still relatively expensive and require costly, specially designed ligands to keep the catalyst in the fluorous phase during the separation. In addition, a significant amount of perfluorinated solvent can remain dissolved in the organic phase, and contamination of the products can occur. To date, there are no industrial developments of this technology, due to lack of competitiveness.
5.3 Ionic Liquids in Multiphasic Reactions |
261 |
|
|
Supercritical carbon dioxide (scCO2) has also emerged as a highly promising reaction medium. In combination with homogeneous catalysis, its benefits could be the potential increase of reaction rates (absence of gas–liquid phase boundary, high diffusion rates) and selectivities, and also its lack of toxicity [9]. In combination with water, it has been used in a biphasic system to perform the hydrogenation of cinnamaldehyde. Gas-liquid-liquid mass transfer limitations were ruled out, due to the very high solubility of reactant gas in scCO2 [10]. Although elegant, this approach still appears relatively expensive, especially for the bulk chemical industry. Furthermore, the low solubility of interesting substrates might hamper the commercialization of scCO2 in the fine chemical industry. A very recent and highly interesting development is the combination of an ionic liquid catalyst phase and a product phase containing scCO2. This approach is presented in more detail in Section 5.4.
Further progress in multiphasic catalysis will rely on the development of alternative techniques that allow the reactivity of a broader range of substrates, the efficient separation of the products, and recovery of the catalyst, while remaining economically viable.
5.3.3
Why Ionic Liquids in Multiphasic Catalysis?
Notwithstanding their very low vapor pressure, their good thermal stability (for thermal decomposition temperatures of several ionic liquids, see [11, 12]) and their wide operating range, the key property of ionic liquids is the potential to tune their physical and chemical properties by variation of the nature of the anions and cations. An illustration of their versatility is given by their exceptional solubility characteristics, which make them good candidates for multiphasic reactions (see Section 5.3.4). Their miscibility with water, for example, depends not only on the hydrophobicity of the cation, but also on the nature of the anion and on the temperature.
N,N’-Dialkylimidazolium cations are of particular interest because they generally give low-melting salts, are more thermally stable than their tetraalkylammonium analogues, and have a wide spectrum of physicochemical properties available. For the same [BMIM]+ cation, for example, the [BF4]–, [CF3SO3]–, [CF3CO2]–, [NO3]–, and halide salts all display complete miscibility with water at 25 °C. On cooling the [BMIM][BF4]/water solution to 4 °C, however, a water-rich phase separates. In a similar way, a change of the [BMIM]+ cation for the longer-chain, more hydrophobic [HMIM]+ (1-hexyl-3-methylimidazolium) cation affords a [BF4]– salt that shows low co-miscibility with water at room temperature. On the other hand, the [BMIM][PF6], [BMIM][SbF6], [BMIM][NTf2] (NTf2 = N(CF3SO2)2), and [BMIM][BR4] ionic liquids show very low miscibility with water, but the shorter, symmetrically substituted [MMIM][PF6] salt becomes water-soluble. One might therefore expect that modification of the alkyl substituents on the imidazolium ring could produce different and very tunable ionic liquid properties.
The influence of the nature of cations and anions on the solubility characteristics of the resulting salts with organic substrates is also discussed in Section 5.3.4. It has

262 Hélène Olivier-Bourbigou, Alain Forestière
1-hexene / IL (wt.%)
|
|
6 |
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
2 |
|
|
|
|
- (CF3 SO2)2N- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-CF3COO- |
|
|
1 |
|
|
|
|
-CF3SO3- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-PF6- |
|
|
|
0 |
|
|
-NO3- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-BF4- |
|
|
N |
+ |
N |
|
|
|
|
|
|
|
N |
+ |
N |
N |
+ |
|
|
|
N |
N |
||||
|
|
|
|
|
|||
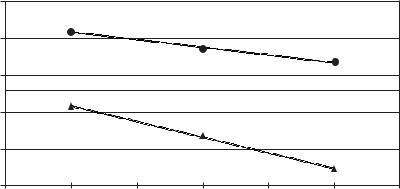
Figure 5.3-1: Solubility of 1-hexene in different ionic liquids as a function of the nature of anions and cations.
been shown (Figure 5.3-1) that increasing the length of the alkyl chain on the imidazolium cation can increase the solubility of 1-hexene, but so can tuning the nature of the anion.
A comparison of the solubility of α-olefins with increasing numbers of carbon atoms in water and in [BMIM][BF4] (Figure 5.3-2), shows that olefins are at least 100 times more soluble in the ionic liquid than in water.
Addition of co-solvents can also change the co-miscibility characteristics of ionic liquids. As an example, the hydrophobic [BMIM][PF6] salt can be completely dissolved in an aqueous ethanol mixture containing between 0.5 and 0.9 mole fraction of ethanol, whereas the ionic liquid itself is only partially miscible with pure water or pure ethanol [13]. The mixing of different salts can also result in systems with modified properties (e.g., conductivity, melting point).
One of the key factors controlling the reaction rate in multiphasic processes (for reactions taking place in the bulk catalyst phase) is the reactant solubility in the catalyst phase. Thanks to their tunable solubility characteristics, the use of ionic liquids as catalyst solvents can be a solution to the extension of aqueous two-phase catalysis to organic substrates presenting a lack of solubility in water, and also to moisture-sensitive reactants and catalysts. With the different examples presented below, we show how ionic liquids can have advantageous effects on reaction rate and on the selectivity of homogeneous catalyzed reactions.
5.3 Ionic Liquids in Multiphasic Reactions |
263 |
|
|
Solubility mmol / L
1000
100
[BMIM][BF4]
10
1
0,1
H2O
0,01
5 |
6 |
7 |
8 |
10 |
Chain length of α-olefin
Figure 5.3-2: Comparison of the solubilities of α-olefins with different numbers of carbon atoms in water and in [BMIM][BF4].
5.3.4
Different Technical Solutions to Catalyst Separation through the Use of Ionic Liquids
In general, homogeneous catalysis suffers from complicated and expensive catalyst separation from the products. Homogeneous catalysts are very often unstable at the high temperatures necessary for the distillation of high-boiling products. Multiphasic catalysis makes the separation of products under mild conditions possible. Different technologies to separate the products, and to recycle the catalytic system when using ionic liquids as one of these phases, have been proposed (Table 5.3-2).
The simplest case (Table 5.3-2, case a) is when the ionic liquid is able to dissolve the catalyst, and displays a partial solubility with the substrates and a poor solubility with the reaction products. Under these conditions, the product upper phase, also containing the unconverted reactants, is removed by simple phase decantation, and the ionic liquid containing the catalyst is recycled. This can be illustrated by transition metal-catalyzed olefin transformations into non-polar hydrocarbon products such as olefin oligomerization, hydrogenation, isomerization, metathesis, and acidic olefin alkylation with isobutane. Transition metal catalysts can also be immobilized in ionic liquids with melting points just above room temperature (Table 5.3-2, case b). The reaction occurs in a two-phase liquid–liquid system. By cooling the reaction mixture, the products can be separated by filtration from the “solid” catalyst medium, which can be recycled. The advantages of this technique have been demonstrated for the hydrogenation of 1-hexene catalyzed by ruthenium-phosphine complexes in [BMIM]Cl/ZnCl2 [14] and for the hydroformylation of 1-hexene in the high-melting phosphonium tosylate ionic liquids [15].

|
5.3 Ionic Liquids in Multiphasic Reactions |
265 |
|||
|
|
|
|
|
|
|
non-polar co-solvent |
||||
unreacted substrate |
|
|
|
products |
|
|
|
|
|||
|
|
|
|
||
reactor |
decantor |
feed |
ionic liquid/catalyst |
heavy by-products
distillation column
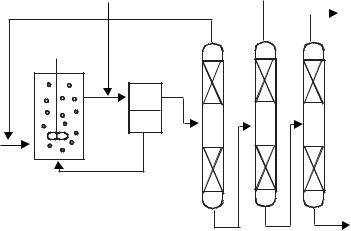
Figure 5.3-3: Example of an extraction method for product separation from ionic liquid/catalyst reaction mixtures.
has the advantage that (i) the catalyst and the products can be separated without heating them, so that thermal deactivation is avoided, and (ii) the extraction is achieved for all the compounds, so that the accumulation of catalyst poisons and high-boiling by-products is minimal. This technology can be applied when ionic liquids are used as the catalyst polar phase (Figure 5.3-3).
A co-solvent that is poorly miscible with ionic liquids but highly miscible with the products can be added in the separation step (after the reaction) to facilitate the product separation. The Pd-mediated Heck coupling of aryl halides or benzoic anhydride with alkenes, for example, can be performed in [BMIM][PF6], the products being extracted with cyclohexane. In this case, water can also be used as an extraction solvent, to remove the salt by-products formed in the reaction [18]. From a practical point of view, the addition of a co-solvent can result in cross-contamina- tion, and it has to be separated from the products in a supplementary step (distillation). More interestingly, unreacted organic reactants themselves (if they have nonpolar character) can be recycled to the separation step and can be used as the extractant co-solvent.
When water-miscible ionic liquids are used as solvents, and when the products are partly or totally soluble in these ionic liquids, the addition of polar solvents, such as water, in a separation step after the reaction can make the ionic liquid more hydrophilic and facilitate the separation of the products from the ionic liquid/water mixture (Table 5.3-2, case e). This concept has been developed by Union Carbide for the hydroformylation of higher alkenes catalyzed by Rh-sulfonated phosphine ligand in the N-methylpyrrolidone (NMP)/water system. Thanks to the presence of NMP, the reaction is performed in one homogeneous phase. After the reaction,