

Organic reaction mechanisms. 1998 / 15
.pdf15 Molecular Rearrangements |
577 |
outlines the proposed mechanism for this transformation which is considered to proceed by the regioselective opening of a transient episulfonium ion.
The observed acid-catalysed conversion of complestatin (289) into chloropeptin L (291) has been envisioned442 as proceeding through a cyclopropyl intermediate (290) (see Scheme 94). An intramolecular oxygen-transfer reaction illustrated in Scheme 95 has been proposed443 to explain hydroxylation of the aromatic nucleus, viz. formation of (292), during the course of a modified Polonovski reaction on galanthamine.
|
|
|
|
|
|
|
|
D |
|
|
|
|
|||
|
HO |
|
|
|
|
|
|
O+ |
H |
D |
|||||
O |
|
H |
|
|
|
|
O |
|
|
|
•• |
|
|
||
|
|
|
|
|
|
|
|
||||||||
|
|
N |
D+ |
|
|
|
|
|
|
N |
|||||
|
|
|
|
|
|
|
|
D |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH |
|
|
|
|
|
|
CH |
|
|
|
CH |
|||
|
|
CH |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
(289) |
|
|
|
|
|
|
|
|
|
|
(290) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
HO |
H |
||
|
N |
|||
O |
|
|
|
|
|
|
|
|
CH |
|
|
|
|
|
|
|
|
||
|
|
D |
||
|
CH |
|
|
|
|
|
(291) |
|
|
SCHEME 94 |
|
|
|
|
Rearrangements Involving Electron-deficient Heteroatoms
An ab initio study of the effects of both substituents and solvents on the Beckmann rearrangement has been undertaken444 and the potential-energy surfaces corresponding to the Beckmann rearrangement of a series of aliphatic and cyclic alkanone oximes have been explored445 using density functional theory. The vapour-phase Beckmann rearrangement of cyclohexanone oxime to -caprolactam, catalysed by mesoporous molecular sieves, has been studied,446 and a weakly acidic borosilicate has also been utilized447 as a catalyst in the above reaction. The non-catalytic Beckmann rearrangement of cyclohexanone oxime to -caprolactam in supercritical water has been
and a comparison has been made of the Beckmann rearrangement of

578 |
Organic Reaction Mechanisms 1998 |
MeO
O |
O |
N |
|
|
Me |
|
OH |
OCOCF3
MeO
|
H |
+O |
N |
Me |
OH
H2O
HO
TFAA
Me
N
O
(292)
MeO

 + OCOCF3 O
+ OCOCF3 O  N
N
Me
OH
|
|
|
|
|
CF3 |
|
MeO |
|
|
|
O |
O |
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
• |
|
|
|
|
|
Me |
•O |
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
|
|||
OH
SCHEME 95
oximes with different molecular sizes over a series of β-zeolites containing different Brønsted acid sites.449 A facile and efficient synthetic procedure for the Beckmann rearrangement of oximes using aluminium chloride in the absence of solvent has been developed,450 and the Beckmann rearrangement of 1-indanone oxime using aluminium chloride has been reported.451 Lactams (294), resulting from the regioselective migration of the C(6)-methylene ring-carbon atom, have been obtained452 from the Beckmann rearrangement of the oximes of 3-phosphonoalkylcyclohexenones (293). A convenient method for the preparation of a bicyclo[3.3.3]undecane derivative via the Beckmann rearrangement of bicyclo[3.3.2]decan-9-one has been described,453 and a novel method for the synthesis of fully protected chiral α, α-disubstituted α-amino
15 Molecular Rearrangements |
|
|
|
|
|
|
|
|
579 |
|||||||||
N |
OH |
|
|
|
|
|
O |
|
|
|
|
|
||||||
|
|
|
|
|
|
H |
|
|
|
|
|
|
||||||
|
|
|
R3 |
|
|
|
|
|
|
|
R3 |
|||||||
|
|
|
|
|
|
N |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PO3Et2 |
|
R2 |
|
|
PO3Et2 |
|
|
|
R2 |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|||||||||||
R1 |
|
|
|
|
|
R1 |
|
|
|
|
R |
|||||||
R |
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
(293) |
|
|
|
|
|
|
(294) |
|
|
|
|
|||||||
pTsO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
N |
O |
|
|
|
|
|
|
|
|
O |
||||||||
|
|
|
|
|
|
propan-2-ol |
|
|
|
|
H |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
Me |
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Me |
|
|
OMe |
|
Et3N |
|
|
|
|
|
|
|
|
OMe |
||||
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
Me |
|
|
|
|
|
|
|
|
Me |
||||||
|
|
|
|
|
|
|
O |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
R |
|
|
|
|
|
|
|
|
|
|
|
R |
|||||
(295) |
|
|
|
|
|
|
|
|
|
|
(296) |
|
|
|
|
|||
acids via a Beckmann rearrangement has been developed454 [see (295) to (296)]. The syntheses of 6-O-methylazithromycin and its aza-ketolide analogue have been achieved455 by carrying out the Beckmann rearrangement of the readily available 9(E)-6-O-methylerythromycin oxime. The reduction of aromatic and cyclic O-(t - butyldimethylsilyl) aldoximes and ketoximes with various reducing agents has been investigated and an attempt has been made to explain the effect of substituents on the novel rearrangement [(297) to (298)] that occurs with a borane–tetrahydrofuran complex.456
A re-evaluation of the Hofmann rearrangement in electron-deficient systems has been undertaken.457 A detailed study of the discrete intermediates, and the sensitivity of the intermediates and products to reagents and to each other in the Hofmann rearrangement of N -α-tosylasparagine, has led to a process that produces 2-(S)- (tosylamino)-β-alanine on a large scale.458
It has been demonstrated that the thermal reaction of a series of alkynylor alkenoylcontaining acyl azides such as (299) involves competition between intramolecular azide cycloaddition and a Curtius rearrangement. Apparently the substituent R plays a key role in determining the competition between the two possible routes.459 It has been shown460 that a silicon group situated at the β-position with respect to an acyl azide group enhances the rate of the Curtius rearrangement by a factor of three, whereas a γ -silyl substituent has a marginal influence. These observations have lent support to the proposition that, during the concerted intramolecular rearrangement of an acyl azide to an isocyanate, an electron-deficient centre at the migration origin is created. An efficient route for the asymmetric synthesis of α, α- disubstituted α-amino acid derivatives, starting from readily available epoxy silyl ethers, has been developed461 using the Curtius rearrangement as a key step (see Scheme 96).
580 |
|
|
|
Organic Reaction Mechanisms 1998 |
||||
|
|
OTBS |
|
|
|
H |
|
|
|
N |
|
|
|
|
|||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
R |
BH3−THF |
|
N |
R |
||
|
|
|||||||
|
|
|
|
|
|
|||
|
|
|
||||||
|
|
|
|
|
|
|
|
|
R′ |
|
R′ |
|
|||||
(297) |
|
(298) |
|
|
||||
COMe
 R
R
N
N3
S
O
(299)
|
R2 |
|
R2 |
OTBS |
|
O |
|
|
|
R1 |
OTBS |
|
R1 |
CHO |
R2 |
CO2H |
|
|
|
|
|
|
|
|
|
R2 |
|
|
OTBS |
||||||
R1 |
NH2 |
|
|
|
|
|
|
|
|
|
R1 |
|
CON3 |
|
|
|||||
|
|
|
|
|
SCHEME 96 |
|
|
|
|
|
|
|
|
|||||||
|
|
Nu− |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
O |
|
|
|
|
|
O |
|
|
|
|
|
|
O |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
N |
|
O OR |
|
|
|
Nu |
|
|
− |
|
O |
|
OR |
|||||||
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
O |
b-alanine |
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
derivatives |
|
|
|
|
Nu |
|
|
N |
|
C |
|
|
O |
|||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
SCHEME 97
The neutral alkali metal salts of benzohydroxamic acids have been found to undergo an unprecedented rearrangement to N ,N -diarylureas.462 A side reaction, producing β-alanine derivatives by way of a Lossen rearrangement, has been observed to accompany the hydrolysis of alkyl succinimidyl carbonates463 in basic aqueous buffers (see Scheme 97). The development of a modified Lossen rearrangement, whereby

15 Molecular Rearrangements |
581 |
N -(t -butyloxycarbonyl)-O-methanesulfonylhydroxamic acids have been converted into protected amines in good yield, has been described464 (see Scheme 98).
A successful synthesis of the tetrahydropyran-protected hydroperoxide (300; 1-18O) using the Baeyer–Villiger strategy has been reported.465 Experimental evidence has been obtained466 to support the fact that, in the Baeyer–Villiger oxidation and Criegee rearrangement, a stereoelectronic effect directs the migratory aptitude, and it is the bond antiperiplanar to the dissociating peroxide bond that always migrates, even when it is electronically disfavoured from doing so.
Me |
Me |
O |
|
|
COCl |
O |
|||||
|
|
|
|
OMs |
|
|
|
|
|
O |
|
Me |
O |
|
|
N |
Et3N, DMAP |
|
|
||||
|
|
|
|
N |
|||||||
|
|
|
|
H |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
MsO |
|
OBut |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
MeCN |
|
PhCH2OH |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
 NHCO2CH2Ph
NHCO2CH2Ph
SCHEME 98
O O
OH
(300)
O = 18O
Rearrangements Involving Organometallic Compounds
An ab initio investigation of the transition state for the Lewis acid-associated migration of an alkyl group from boron to an α-dichloro-carbon in a non-racemic boronic ester has been carried out.467 The calculated transition state has shown that it is important to have the non-participating chlorine atom anti to the metal, e.g. as in (301). The
|
R |
|
|
R |
|
|
O − |
||
|
|
|
||
|
|
O |
B |
H |
|
|
|
Cl |
|
|
|
|
|
|
R |
|
M |
Cl |
|
|
Cl |
|
Cl |
|
|
|
|
|
|
|
|
(301) |
|
|
582 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Organic Reaction Mechanisms 1998 |
|||||||||||||
|
OEt |
|
|
|
|
|
|
|
|
OEt |
|
|
|
|
|
|
|
|
|
|
M(CO)5 |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(CO)5M |
Ph |
|
|
|
|
(CO)5M |
|
|
|
|
|
Ph |
|
|
|
|
EtO |
+ |
|
|
• |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
R2N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
||||
|
|
|
|
|
|
R2N |
|
H |
|
|
|
|
|
|
|
|
H |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
O |
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
(302) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
M = W, Cr |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OEt |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
(CO)5M |
|
|
|
|
|
|
Ph |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
R2 |
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
R2N |
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
EtO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
|||||||
|
|
|
|
|
|
|
|
OEt |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
(CO)5M |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
O |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
(304) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(303) |
||||
SCHEME 99
stereoselectivity is then dictated by placing the metal on the least-hindered side of the oxygen, trans to the R group of the ester. This combination places the Lewis acid in the sterically least hindered position.
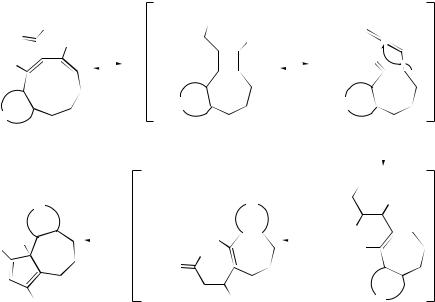
2-Aminocyclonona-1,8-dienyl carbene complexes (302) in solution have been found to undergo ring contraction of the nine-membered ring to give (2-amino- cycloheptenyl)alkenyl carbene complexes (303) which are subsequently transformed into tetrahydroazulenes (304) by elimination of the metal unit468 (see Scheme 99), and (cyclobutenyl)carbene tungsten complexes (305) have been shown to rearrange
to 1-tungstahexa-1,3,5-trienes (306) by ring |
opening |
of |
the cyclobutene |
ring |
and subsequent [1,3]-hydrogen migration.469 |
It has |
been |
reported470 that |
the |
d2[{p-But -calix[4]-(O4)}W] fragment assists a variety of ethylene rearrangements
which |
are very similar |
to those often supposed to occur on |
metal |
oxides. |
Such |
rearrangements are |
driven by light, acids, and bases, or |
occur |
under |
reducing conditions. Quantum-mechanical calculations have shown that471 that two energetically nearly degenerate pathways are possible for the rearrangement of tungsten–acetylene complexes (307) to the energetically higher lying vinylidene complexes (310). The direct [1,2]-hydrogen migration was found to proceed via a transition state (308) which has a non-planar C2H2 moiety. The alternative pathway involves the alkynyl(hydrido)metal complex (309). Complexes of the chiral bis(oxazoline) 2,6-bis[(4S)-isopropyloxazoline-2-yl]pyridine (311; M = Mo or W), in which the ligand is restricted to a bidentate bonding mode, have been found to be

15 Molecular Rearrangements |
583 |
|
|
|
OEt |
||||||
(CO)5W |
|
|
|
|
Ph |
||||
|
|
|
|
||||||
|
|
|
|||||||
R |
|
|
N |
|
|
R′ |
|||
|
|
|
|
||||||
|
|
|
|
||||||
H |
|
|
|
|
OEt |
||||
|
Ph |
|
|
|
|
|
W(CO)5 |
||
|
|
|
|
|
|
||||
|
|
|
|
|
|
||||
|
|
|
|
|
|
||||
|
|
|
|
EtO |
|||||
|
|
|
(305) |
|
|
|
|
||
 H
H
H  LnM C
LnM C 
C
|
|
|
C |
(308) |
||||
|
|
|
|
|
|
|||
LnM |
|
|
|
|||||
C |
|
|
H |
|||||
|
|
|
|
|
||||
|
|
|
|
|
|
LnM |
|
|
|
|
H |
|
M |
||||
|
|
|
||||||
(307) |
|
|
|
|
|
|
C |
|
|
|
|
|
(309) |
||||
|
|
|
|
|
|
|||
|
|
|
|
|
OEt |
|
|
|
|
|
|
|
||
(CO)5W |
|
|
|
Ph |
|
R′ |
||||||||
|
|
|
|
|
||||||||||
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
||||||
|
R |
N |
|
|
||||||||||
|
|
|||||||||||||
|
H |
+ |
|
|
|
|
|
|
||||||
|
Ph |
OEt |
|
|
|
|||||||||
|
|
|
|
|
• |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
− |
|
|
|
|||
|
|
|
EtO |
W(CO)5 |
||||||||||
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
−H+ |
|
+H+ |
|
|
|
||||
|
|
|
|
|
OEt |
|
|
|
|
|
|
|
||
(CO)5W |
|
|
|
|
|
Ph |
R′ |
|||||||
|
|
|
|
|
||||||||||
|
|
|||||||||||||
|
|
|
|
|
|
|
|
N |
|
|
||||
|
R |
|
|
|||||||||||
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
OEt |
|
|
|
|||||||||
 H
H
EtO
W(CO)5
(306)
H |
H |
LnM  C
C  C
C
H
(310)
C
H
O
O
N |
L |
N |
|
• |
|||
• |
• |
||
• |
|
|
• |
• |
|
|
• |
• |
|
|
|
• |
|
|
• |
• |
|
|
• |
• |
|
• |
|
• |
• |
|
|
|
|
• |
|
N• • • • • • • • • • M
CO
OC
CO
CHMe2
(311)
CHMe2

584 |
Organic Reaction Mechanisms 1998 |
fluxional, with exchange occurring between the pendant and coordinated oxazoline rings. The energetics and mechanism of the rearrangement have been studied in detail472 by oneand two-dimensional NMR techniques.
Reduction of (312) has been found to afford the dimer (313) which upon heating rearranged to yield the unprecedented di(benzopentalene) complex (314). The regioand stereo-specificity of the conversion (313) into (314) implies a metalmediated pathway for the process473 (see Scheme 100). The first observable cis- bis(alkyne)cyclobutadiene rearrangement [see (315) to (316)] has been reported.474
Mn+ |
BF4− |
CO CO CO |
|
(312) |
|
CO
OC CO
Mn
Mn

OC  CO
CO
CO
(314) |
|
|
CO |
Me |
|
|
||
OC |
|
|
Re |
Me |
|
Me |
||
OC |
||
|
||
CO |
|
|
|
Me |
|
(315) |
|
CO
OC CO
Mn
Mn
OC CO
CO
(313)
H Mn
Mn
OC CO
CO
SCHEME 100
CO |
Me |
|
Me |
||
OC |
||
|
||
Re |
Me |
|
OC |
||
Me |
||
CO |
||
|
(316) |

15 Molecular Rearrangements |
|
|
|
|
|
585 |
|
The binuclear iron complexes (317) |
have |
been |
found |
to |
undergo a |
||
thermal |
rearrangement475,476 to afford |
the complexes |
(318), which were |
||||
|
|
|
|
and Fe Fe bonds in |
|||
evidently |
formed via a metathesis |
between |
Si−Si 477 |
|
− |
|
|
(317). A similar rearrangement has been observed |
in a disilyl-bridged |
||||||
bis(cyclopentadienyl)tetracarbonyldiruthenium complex. Unprecedented |
and stable |
||||||
(η − η6 : η6-pentafulvadiene)diruthenium |
complexes (320) |
have |
been |
prepared478 |
|||
from a two-electron oxidation of trans-1,2-bis(ruthenocenyl)ethylenes (319), and dimethyl analogues have been similarly obtained from trans- and cis-1,2-dimethyl- 1,2-bis(ruthenocenyl)ethylenes.
R |
Me2 |
|
Me2 |
|
Si |
|
Si |
R1 |
|
|
|
|
R |
O |
R |
R |
|
|
|
|
C |
|
|
|
|
|
|
|
Fe |
|
Fe |
|
|
||
|
OC |
C R2 |
|
|
|
O |
|
(317)
R, R1 = Me; R2 = CO
R = H; R1 = But; R2 = P(OPh)3
Me
Me 
 Me
Me
Me Ru Me
Ru
Me
Me
Me
(319)
R
 R1
R1
R
−2e
Me +2e
Me
CO
Me2
RSi Fe
Fe R2
R2
|
|
|
R |
R1 |
R |
R1 |
|
|
|
R |
|
|
R |
|
|
|
OC Fe Si |
R |
|
|
|
Me2 |
|
|
|
CO |
|
|
(318) |
|
|
Me
Me 
 Me
Me
Me Ru Me

 Me Ru Me
Me Ru Me
Me Me
Me
(320)
A hitherto unknown type of rearrangement of 1-(1-alkynyl)cyclopropanols (321) to cyclopent-2-en-1-ones (322) mediated by octacarbonyldicobalt479 and hexacarbonyldicobalt480 complexes has been described. A possible pathway for the transformation is outlined in Scheme 101. A β-proton transfer accompanied by a metal-mediated Stevens rearrangement, which converts a coordinated dimethylsulfane

586 |
|
|
Organic Reaction Mechanisms 1998 |
|||||
|
|
Ph |
Ph |
|
||||
|
|
|
|
|
|
|
|
|
|
HO |
|
|
OC |
C |
CO |
||
|
|
|
OC |
|
Co |
|
Co |
|
|
|
|
|
|
|
|||
|
|
Co2(CO)6 |
C |
CO |
||||
|
|
|
|
OC |
||||
|
|
(321) |
|
|
|
|
HO |
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
O |
|
|
|
|
|
|
|
Ph |
|
L |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OC |
C |
|
|||
|
|
|
−[Co2(CO)5L] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Co |
CO |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OC |
|
Co |
|
CO |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
(CO)3Co(CO)2LCo |
|
|
|
|
|
OC |
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
C |
|
|
||||||||||||||
Ph |
|
|
|
|
Ph |
|
|
|
|
|
|
|
H |
|
|
|||||||||
(322) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
||
|
|
|
|
|
|
|
|
SCHEME 101 |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
CF3 |
|
|
|
|
CF3 |
CO |
|
|
|||||||||||
|
|
|
F3C |
|
|
|
|
|
|
|
|
|
F3C |
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
RhCp |
|
|
||||||
|
|
|
|
|
|
|
Me2S |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
RhCp |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
CpRh |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
CpRh |
|
|
|
C |
|
|
S•• |
|
H |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
O |
Me |
CH2 |
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
−CO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
CF3 |
|
|
|
|
|
CF3 |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
F3C |
|
|
|
F3C |
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
H + |
|
|
|||||||||
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
CpRh |
|
RhCp |
|
|
|||||||||||
|
|
|
|
|
|
|
|
RhCp |
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
CpRh |
|
|
S•• − |
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
S |
Me |
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2 |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
CH2Me
SCHEME 102
to a bridging ethanethiolate group (see Scheme 102), has been invoked481 to account for the observed rearrangement of organic chalcogenides on a rhodium–rhodium bond. The iridium-catalysed isomerization of allyl silyl ethers has been rationalized482 as proceeding through the oxidative addition of an allylic C−H bond to the iridium(I) metal centre, giving a syn-π -allyliridium complex (323) which selectively leads to the E-isomer (see Scheme 103).