

Дизостоз
Дизостоз (dysostosis; диз- + греч. osteon кость + -оз) — общее название аномалий развития костей скелета, лежащих в основе семейных наследственных болезней костной системы.
Дизостоз клейдокраниальный (d. cleidocranialis; греч. kleis, kleidos ключ, ключица + kranion череп) — см. Дизостоз ключично-черепной.
Дизостоз ключично-черепной (d. claviculocranialis; син.: Д. клейдокраниальный, Шейтхауэра — Мари — Сентона синдром) — Д., характеризующийся незаращением родничков черепа, брахицефалией, гипоплазией лицевых костей (главным образом верхней челюсти), полным или частичным недоразвитием ключиц.
Дизостоз множественный (d. multiplex) — см. Гаргоилизм.
Дизостоз ротопальцелицевой (d. orodigitofacialis; син.
Папийон-Леаж — Псома синдром) — Д., характеризующийся узким («орлиным») носом, укорочением средней части верхней губы, расщеплением языка и верхнего неба, нарушением развития зубов, недоразвитием фаланг пальцев и т.д.; наследуется по аутосомно-доминантному типу.
Дизостоз челюстно-лицевой (d. mandibulofacialis; син. Франческетти — Цвалена синдром) — Д., характеризующийся гипоплазией нижней челюсти и скуловых костей с нарушениями развития зубов,
деформацией ушных раковин, а иногда и среднего уха, а также макростомией («рыбье» или «птичье» лицо); наследуется по аутосомно-доминантному типу.
Дизостоз челюстно-черепной (d. maxillocranialis; син. Петерс — Хевельса синдром) — Д., характеризующийся гипоплазией верхней челюсти, скуловых дуг, прогенией, укорочением переднего отдела основания черепа; наследуется по аутосомно-доминантному типу.
Дизостоз черепно-лицевой (d.
craniofacialis; син. Крузона синдром) — Д., характеризующийся сочетанием недоразвития костей черепа с преждевременным закрытием черепных швов, гипертелоризмом, экзофтальмом, косоглазием, расстройством зрения, крючковатой формой носа («клюв попугая»); наследуется по аутосомно-доминантному типу.
Дизостоз энхондральный политопный (d. polytopica enchondralis) — см.Лери — Вейлля дисхондростеоз.
20. Аутосомно - рецессивный тип наследования
При
аутосомно-рецессивных заболеваниях у
здоровых родителей некоторые дети
оказываются больными. Это связано с
тем, что оба родителя являются носителями
аномального аутосомно-рецесссивного
гена. Для этого типа наследования
характерны следующие закономерности.
1)
если поражённый ребёнок родился у
фенотипически нормальных родителей,
то родители обязательно являются
гетерозиготами, 25% их детей поражено,50%
- гетерозиготны и 25% - нормальны.
2) Если
вступают в брак больной с рецессивным
заболеванием и генотипически нормальный
человек, все их дети будут гетерозиготами
и фенотипически здоровы.
3) Если вступают
в брак больной и гетерозигота, то половина
их детей окажутся поражёнными, а половина
– гетерозиготны.
4) Если вступают в
брак двое больных одним и тем же
рецессивным заболеванием, то все дети
их будут больны.
5) Мужчины и женщины
болеют одинаково часто.
|
|
Фенилкетонурия впервые
была открыта учёным-генетиком Феллингом
в 1934 году. В основе её развития лежит
резкое снижение активности фермента,
превращающего аминокислоту фенилаланин
в тирозин. У гомозиготных по этой мутации
людей в крови значительно повышается
концентрация фенилаланина и ряда других
соединений, оказывающих вредное
воздействие. В итоге гомозиготные дети
отстают в развитии, у них наблюдаются
повреждения мозга и возникает умственная
отсталость. В среднем один человек из
50 гетерозиготен по гену фенилкетонурии.
Особенность фенилкетонурии в том, что
она проявляется на первом году жизни
ребёнка и медленно прогрессирует. Без
рано начатого лечения это заболевание
делает ребёнка инвалидом на всю жизнь,
так как вызывает отравление клеток
мозга (интоксикация мозга).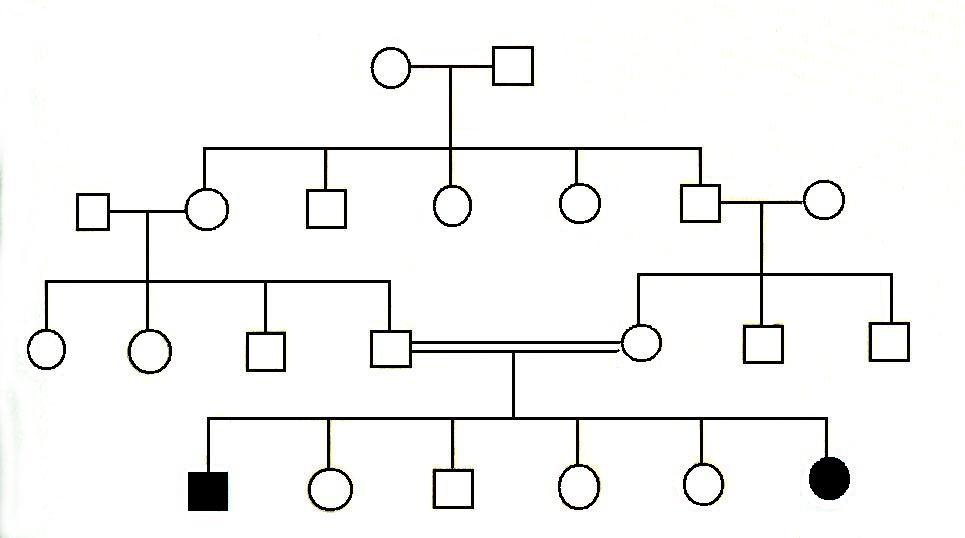
|
|
Синдром
Элерса — Данлоса
Вызывает
недостаточное развитие коллагеновых
структур в различных системах организма.
Проявляется патологией кожи,
опорно-двигательного аппарата,
сердечно-сосудистой системы, глаз,
гиперрастяжимостью кожи — взятая в
складку кожа легко оттягивается. При
этом оттянутая кожная складка быстро
возвращается в исходное положение. Кожа
тонкая, нежная, бархатистая на ощупь,
слабо фиксирована с подлежащими тканями.
Мышечно-скелетные включают деформации
грудной клетки, кифоз, сколиоз, косолапость.
Зубы у больных могут быть неправильно
сформированы, аномально расположены,
уменьшены в размерах или частично
отсутствуют. Умственное развитие больных
в большинстве случаев соответствует
возрасту.
|
|
Муковисцидоз
Характеризуется
системным поражением экзокринных желез
и проявляется тяжелыми расстройствами
функций органов дыхания, желудочно-кишечного
тракта и ряда других органов и систем.
Частота муковисцидоза в по данным разных
авторов, составляет от 1:2500 до 1:8000
новорожденных. Заболевание наследуется
по аутосомно-рецессивному типу,
характеризуется кистозным перерождением
поджелудочной железы, желез кишечника
и дыхательных путей из-за закупорки их
выводных протоков вязким секретом;
проявляется в форме хронической
пневмонии, расстройств пищеварения.
Фенилкетонурия, фенилпировиноградная олигофрения, наследственное заболевание из группы ферментопатий, в основе которого лежит аномалия аминокислотного обмена вследствие отсутствия или резкого снижения активности фермента фенилаланингидроксилазы. Описана в 1934 норв. учёным А. Фёллингом (A. Foiling) (болезнь Фёллинга). Частота Ф. – 1 случай на 10–15 тыс. новорождённых; наследуется по аутосомно-рецессивному типу (см.Наследственные заболевания). При Ф. фенилаланингидроксилаза сохраняет только около 5% активности, в связи с чем нарушается обмен фенилаланина и вследствие этого – тирозина, триптофана и др., накапливаются промежуточные продукты обмена – фенилэтиламин, фенилпировиноградная кислота и др. и возникает дефицит метаболитов, необходимых для нормального функционирования организма В частности, вероятная причина умственных расстройств – дефицит медиаторов нервной системы (адреналина, норадреналина, серотонина и др.). Т. о., при Ф. возникает комплекс взаимосвязанных метаболических расстройств, состоящий из первичного ферментного нарушения и обусловленных им др. нарушений обмена.
Ф. проявляется главным образом выраженной олигофренией (идиотией или имбецильностью). Диагностируется в первые дни жизни ребёнка с помощью экспресс-методов – микробиологических или биохимических. Последние основаны на определении пировиноградной кислоты в моче посредством индикаторов (проба Фёллинга). Лечение сводится главным образом к специальной диете (резкое ограничение продуктов, содержащих фенилаланин)
Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия,гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова) — врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов.
Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передается по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме (участок 13q14-q21).
Болезнь Тея — Сакса (ранняя детская идиотия амавротическая) — редкое наследственное заболевание нервной системы. Названо в честь британского офтальмолога Уоррена Тея(англ. Warren Tay, 1843—1927), и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944).
|
Содержание [убрать]
|