

патофизиология_1-680 Книга
.pdf(«корнеальная дуга»). Возможно развитие аортального стеноза из-за ксантоматоза полулунных клапанов.
Увеличение содержания в крови ЛПНП может возникать при печеночной недостаточности, нефротическом синдроме, гиперкортицизме, гипотиреозе, хроническом стрессе и других формах патологии.
Подтип IIб характеризуется одновременным повышением содержания ЛПНП и ЛПОНП. Первичная гиперлипопротеинемия IIб типа в большинстве случаев имеет в своей основе наследственное заболевание семейную смешанную гиперлипидемию. У таких пациентов имеется генетический дефект рецептора ЛП, в результате которого нарушается связывание и ЛПНП, и ЛПОНП. В отличие от типа IIа повышены и уровень ХН, и концентрация ТГ. Рано развивается атеросклероз.
Вторичные формы II типа (IIа и IIб) возникают при гиперкортицизме, пищевой перегрузке ХН и ТГ, перемежающейся порфирии, диабете, у больных с нефротическим синдромом, с гипофизарной карликовостью, обусловленной изолированным дефицитом СТГ.
III тип — диз-бета-липопротеинемия. Эта ГЛП в первичном варианте известна как болезнь устранения остаточных частиц, или болезнь широкой β-полосы. Заболевание поражает гомозигот по одному из мутантных аллелей апопротеина Е. Экспрессия генов облегчается при пищевой перегрузке ХН и ТГ. При этом типе повышено содержание липопротеидов промежуточной плотности (ЛППП), определяются остаточные хиломикроны, отмечается высокая степень поражения атеросклеротическим процессом всего сосудистого русла, даже периферических сосудов. Клинические признаки атеросклероза проявляются после 20 лет поражением сердечно-сосудистой системы.
Приобретенные формы имеют место при холестазе, гипотиреозе, моноклональной гаммапатии.
IV тип — гиперлипопротеинемия — это самая распространенная ДЛП. Наследственный вариант известен как семейная триглицеридемия с аутосомнодоминантным типом наследования. Проявляется после полового созревания. Патогенез заболевания связан либо с повышением синтеза апопротеина В, либо первичной гиперпродукцией ЛПОНП в печени.
Нарушение спектра ЛП по этому типу приобретенного характера наблюдается при алкоголизме, хроническом стрессе, остром гепатите, уремии, акромегалии, гликогенозе I типа фон Гирке, диабете, липодистрофиях, моноклональных гаммапатиях, применении пероральных контрацептивов и β-адреноблокаторов, гемодиализе.
V тип — гиперхиломикронемия и гипер-бета-липопротеидемия (первичная гиперлипидемия V типа) — развивается при аутосомно-рецессивном отсутствии апопротеина С II, то есть кофактора липопротеиновой липазы (ЛПЛ). В крови накапливаются ХМ и ЛПОНП (комбинация I и IV типов ГЛП).
Вторичная ГЛП этого типа бывает при гликогенозе Гирке, алкоголизме и использовании противозачаточных пероральных средств. Возможно вторичное торможение активности ЛПЛ избытком ХМ.
Некоторые аномалии спектра ЛП (дислипопротеинемии) не вошли в эту классификацию. К ним относятся появление липопротеина (а), гипоальфапротеинемия, анальфапротеинемия, наследственная недостаточность лецитинхолестеринацилтрансферазы, церебральный ксантоматоз, ситостеринемия и некоторые дру-
431
гие варианты патологии, ускоряющие развитие атеросклероза. Из перечисленных ДЛП заслуживает особого внимания появление минорного ЛП — липопротеин (а), чрезвычайно повышающего риск развития атеросклероза. Липопротеин (а) рассматривается как структурный аналог и ингибитор плазминогена и фибринолиза, индуктор пролиферации ГМК, активатор тромбоцитов и свертывания крови, поэтому риск атеросклероза и тромбоза увеличивается даже при невысоком общем ХН. 90 % лиц с нетипичными формами ИБС и 25 % всех лиц с ИБС моложе 60 лет имеют повышение уровня этого минорного ЛП.
Следует отметить, что ГЛП с повышением общего и свободного ХН гораздо атерогеннее,чемте,прикоторыхповышаетсятолькоуровеньТГ.ЭХнеатерогенны. Патологические качественные модификации липопротеидных частиц — окисление, гликозилирование, ацетилирование, обогащение лизолецитином и свободным ХН, присоединение к апопротеинам аутоантител значительно усиливают атерогенность ЛП, так как способ их взаимодействия с клетками сосуда при этом меняется. Атеросклероз ускоряется при недостатке полиеновых и ω-3-ненасыщенных жирных кислот и антиоксидантов в ЛП.
17.5.4. Морфологические проявления и стадии развития атеросклероза
Современная классификация атеросклеротических поражений у человека, разработанная в 1995 году Stary и принятая кардиологическими обществами ряда стран, включает:
Тип I — начальные поражения, характеризуются изменениями в эндотелии, в частности, накоплением богатых холестерином липопротеиновых частиц и наличием отдельных пенистых клеток макрофагального происхождения.
Тип II — липидные пятна и полоски, характеризуются преимущественно внутриклеточным депонированием липидов в скоплениях пенистых клеток макрофагального и гладкомышечного генеза.
Липидные пятна представляют собой желтоватые точки диаметром до 1,5 мм, мягкой консистенции, не возвышающиеся над поверхностью эндотелия и не создающие препятствий току крови. Липидные пятна раньше всего появляются в аорте, в 10-летнем возрасте липидные пятна занимают около 10 %, к 25 годам — до 30–50 % внутренней поверхности аорты, к 15 годам — липидные пятна появляются в коронарных артериях, а к 35–45 годам — в церебральных артериях.
Липидные полоски — следующая ранняя стадия развития атеросклеротического поражения из липидных пятен, которые увеличиваются в размерах, становятся удлиненными (до 15 мм длиной) и более широкими (до 3 мм в диаметре). Они состоят, как и липидные пятна, из пенистых клеток макрофагального и миоцитарного происхождения, нагруженных липидами , и Т-лимфоцитов. Наблюдается пролиферация гладкомышечных клеток по периферии атеросклеротического поражения.
Тип III — переходные поражения, сходные с II типом, но отличающиеся некоторым количеством внеклеточных липидных депозитов в виде эфиров холестерина и свободного холестерина.
432
Тип IV — атеромы, располагают значительным ядром внеклеточных липидов. Тип V — фиброатеромы, отличаются наличием фиброзной «крышки» над липидным ядром, могут кальцифицироваться или бывают преимущественно фи-
брозными.
Липидное ядро является центральной частью фиброатеромы и представляет собой аморфную массу, состоящую из эфиров холестерина, кристаллов свободного холестерина, продуктов распада эластических и коллагеновых волокон, пенистых клеток (по периферии ядра), Т-лимфоцитов, могут встречаться плазматические клетки. Липидное ядро покрыто фиброзной покрышкой (капсулой), которая образуется вследствие секреции компонентов экстрацеллюлярного матрикса пролифирирующими и мигрирующими в интиму ГМК. Степень выраженности фиброзных изменений зависит от типа пенистых клеток. Если пенистые клетки преимущественно макрофагального происхождения, то количество внеклеточных липидов в ней велико, но фиброзная покрышка сравнительно тонкая. Такие бляшки называют «желтыми». Если пенистые клетки имеют миоцитарный генез, липидное ядро несколько меньше, совершенно явно преобладают фиброзные изменения, фиброзная оболочка хорошо выражена, плотна, и такая бляшка называется фиброзной или «белой». Она вызывает гемодинамически значимое сужение артерии.
ТипVI — осложненные фиброатеромы, имеются поверхностные дефекты, кровоизлияния в бляшку, вторичное тромбобразование, атеромы проникают в медию.
Атеросклероз охватывает в наибольшей степени ряд артериальных сосудов, в которых сильнее всего выражена нагрузка на стенку. В первую очередь, это абдоминальная аорта, находящаяся «между молотом пульса и наковальней позвоночника» (В. Кумар и соавт., 1997).
17.5.5. Механизм развития атеросклеротических поражений сосудов
Атеросклеротическое поражение артерий начинается с накопления малых липопротеиновых частиц, богатых холестерином, в интиме артерий. Аккумуляция липопротеиновых частиц обусловлена, с одной стороны, повышенной проницаемостью эндотелия, с другой стороны, связыванием липопротеиновых частиц с компонентами экстрацеллюлярного матрикса, прежде всего с молекулами одного из протеогликанов — гепарансульфата.
Проникновению липопротеинов в интиму артерий способствуют повреждение эндотелия или его дисфункция, проявляющаяся повышением проницаемости и адгезивности, увеличением секреции прокоагулянтов и сосудосуживающих веществ. Факторами, вызывающими дисфункцию эндотелия артерий, могут быть артериальная гипертензия; изменения гемодинамики (гидравлический удар потока крови по эндотелиальному слою в местах бифуркации, изгибов, сужений, ветвлений артерий); значительное увеличение содержания в крови липопротеинов, богатых холестерином; курение, гипоксия и гипоксемия различного генеза; высокий уровень в крови гомоцистеина, обладающего высокой эндотелиотоксичностью; вирусная инфекция и другие. Эти факторы приводят к разрыхлению и истончению защитного гликокаликса на поверхности эндотелиальных клеток, расширению межэндотелиальных щелей, отеку субэндотелиального слоя интимы, разъединению его клеток и волокнистых структур.
433
В норме межэндотелиальные промежутки очень узки и непроницаемы для липопротеинов. Однако под влиянием поступления в кровь вазоактивных веществ
—катехоламинов, ангиотензина II, серотонина, эндотелина и других, а также под воздействием выраженной гиперхолестеринемии межэндотелиальные промежутки раскрываются, и частицы ЛПНП проникают в интиму артерий.
Липопротеиновые частицы, проникшие в экстрацеллюлярное пространство интимы и связанные с протеогликанами, подвергаются модификации . К модифицированным липопротеинам относятся гликозилированные ЛП, перекисномодифицированные ЛПНП, аутоиммунные комплексы ЛП-антитело, продукты ограниченного протеолиза ЛП, десиалированные ЛПНП, комплексы ЛПНП с гликозаминогликанами, агрегированные ЛП. Модификация липопротеинов происходит не только в интиме артерий, но и в крови. Наибольшее значение в развитии атеросклероза имеют такие разновидности химической модификации ЛПНП, как гликозилирование и пероксидация.
Окисленные ЛПНП стимулируют продукцию эндотелиоцитами гемоаттрактантных белков для лейкоцитов. Взаимодействию лейкоцитов с эндотелиальными клетками и последующему их проникновению в субэндогелиальное пространство предшествует адгезия этих клеток к поверхности эндотелия. Адгезия происходит при участии специальных адгезивных молекул и некоторых цитокинов. В разви тии атеросклероза принимают участие две группы молекул адгезии — селектины и молекулы суперсемейства иммуноглобулинов. Селектины Р-селектин, Е-селектин и L-селектин представляют собой семейство Са2+-зависимых трансмембранных лектинов первого типа, экспрессированных на эндотелиоцитах и лейкоцитах, и включает
Р-селектин постоянно синтезируется эндотелиоцитами и содержится также в гранулах тромбоцитов. К Р-селектину на поверхности лейкоцитов — моноцитов, лимфоцитов, гранулоцитов имеются соответствующие лиганды-гликопротеины
(PSGL-1 — P-selectin glycoprotein ligand-1). Именно Р-селектины принимают наи-
большее участие в адгезии лейкоцитов.
Е-селектин в обычных условиях не экспрессируется на эндотелиоцитах, но при действии ИЛ-1, ФНОα и интерферона-β начинается его синтез, и он появляется на поверхности эндотелиоцитов. Лигандами для Е-селектина являются сиализированные олигосахариды — сиалил Льюис X, которые расположены преимуще ственно на гранулоцитах и в меньшем количестве на моноцитах и Т-лимфоцитах
—клетках памяти. Е-селектины, таким образом, способствуют адгезии и скоплению преимущественно полиморфноядерных лейкоцитов и, главным образом, на стадии раннего развития атеромы. Наибольшее значение Е-селектин имеет в развитии воспалительного поражения сосудов.
L-селектин экспрессирован практически на всех лейкоцитах, но не экспрессирован на эндотелиоцитах. На поверхности эндотелиоцитов имеются лиганды к
L-селектину: MadCAM-1 — (mucosal addressin cell adhesion molecule-1); PSGL-1; GlyCAM-1 (glycosylation-dependent cell adhesion molecule-1); CD-34; Sgp 200 (sulfated glycoprotein p200).
Большую роль в адгезии лейкоцитов к эндотелию играют адгезивные молекулы VCAM-1 и ICAM-1, относящиеся к суперсемейству иммуноглобулинов.
434
VCAM-1 (vascular cell adhesion molecules, адгезивные молекулы сосудистой стенки) продуцируются только эндотелиальными клетками. Эти адгезивные молекулы взаимодействуют со специфическими лигандами с интегринами VLA-4 (very late antigen-4), расположенными на поверхности моноцитов и Т-лимфоцитов. Молекулы адгезии VCAM-1 принимают участие в развитии ранних стадий атеросклеротического поражения. Лизофосфатидилхолин, входящий в состав окислительно модифицированныхЛПНП,можетувеличиватьэкспрессиюVCAM-1.Ламинарный ток крови в нормальных сосудах подавляет экспрессию VCAM-1, что обусловлено влиянием азота оксида, продуцируемого эндотелием неповрежденных сосудов. Нарушение ламинарного кровотока в артериях сопровождается снижением синтеза ферментов супероксиддисмутазы и NO-синтазы. Как известно, супероксиддисмутаза ингибирует активность перекисного окисления липидов, а под влиянием NOсинтазы образуется вазодилатирующий фактор — азота оксид.
ICAМ-1 (intercellular adhesion molecules, молекулы межклеточной адгезии)
вырабатываются не только эндотелиальными клетками, но и активированными гладкомышечными клетками и макрофагами. Эти адгезивные молекулы взаимо-
действуют с лигандами CD 11а integrin (LFA-1) и CD 11b integrin (Mac-1), располо-
женными на всех типах лейкоцитов. Цитокины ИЛ-1 и ФНО увеличивают экспрес-
сию ICAM-1 и VCAM-1.
Адгезия лейкоцитов к эндотелиоцитам является стадийным процессом и включает привлечение лейкоцитов к эндотелию из крови и образование слоя лейкоцитов, перекатывание их по поверхности эндотелия , плотное прилипание лейкоцитов к эндотелию и затем их миграцию в субэндотелиальное пространство интимы.
Важнейшими лейкоцитарными клетками, проникающими через межэндотелиальные промежутки, являются моноциты. Миграция лейкоцитов в субэндотелиальное пространство происходит под влиянием хемоаттрактантных цитокинов или хемокинов, а также под влиянием модифицированных липопротеиновых частиц низкой плотности.
Поступившие и скопившиеся в субэндотелиальном пространстве интимы моноциты превращаются в макрофаги, которые поглощают модифицированные ЛПНП с помощью скэвенджер-рецепторов и превращаются в пенистые клетки. Дифференциация моноцитов, пролиферация макрофагов и экспрессия на их поверхности скэвенджер-рецепторов индуцируются моноцитарным колониестимулирующим фактором (M-CSF).
Макрофагимогутсвязыватьиподвергатьраспадукакнеизмененные(нативные), так и модифицированные ЛПНП. Нативные ЛПНП захватываются макрофагами с помощью апо-В, Е-рецепторов, однако при этом не происходит накопления эфиров холестерина в макрофагах. Это объясняется функционированием механизма своеобразной обратной регуляции. При накоплении в макрофаге избытка холестерина угнетаетсясинтезапоВ,Е-рецепторов,чтоведеткуменьшениюсвязыванияизахвата новых частиц нативных ЛПНП.
Совсем иначе ведут себя скэвенджер-рецепторы макрофагов, связывающие модифицированные ЛПНП. Накопление холестерина за счет модифицированных ЛПНП приводит к перегрузке макрофагов холестерином и превращению их в пенистые клетки. Таким образом, именно модифицированные ЛПНП являются атерогенными.
435
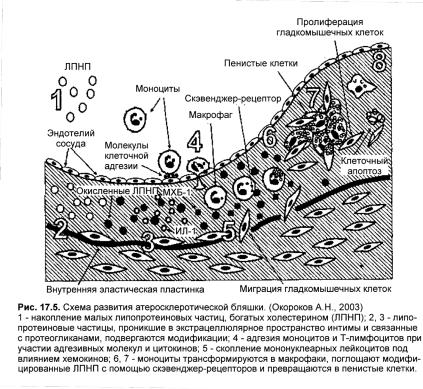
Пенистые клетки, т.е. макрофаги, перегруженные липидами, в большинстве своем остаются в интиме артерий и погибают, подвергаясь апоптозу — запрограммированной клеточной смерти. При этом происходит выделение накопленных в пенистых клетках эфиров холестерина, неэстерифицированного холестерина и кристаллов моногидрата холестерина . Эти процессы приводят к очаговым скоплениям холестерина в интиме артерий и создают предпосылки для развития липидных пятен, затем липидных полосок и в последующем атеросклеротических бляшек. Пенистые клетки являются также источником ряда цитокинов и эффекторных молекул, таких как супероксидный анион кислорода О2~ и металлопротеиназы матрикса, имеющих значение в развитии и прогрессировании атеросклеротических поражений.
Дальнейшее развитие атеросклеротического поражения характеризуется миграцией в интиму гладкомышечных клеток и их пролиферацией.
В норме гладкомышечные клетки располагаются в tunica media (средней оболочке артерий) и выполняют сократительную функцию. Гладкомышечные клетки мигрируют в интиму под влиянием хемоаттрактантов, которые продуцируются макрофагами, эндотелиоцитами, фибробластами интимы артерий в ответ на появление в ней модифицированных ЛПНП. Наибольшее значение для миграции
436
гладкомышечных клеток в интиму артерий имеет тромбоцитарный фактор роста
(platelet-derived growth factor, PDGF), который секретируется не только тромбо-
цитами (они также могут проникать в интиму из крови), но и эндотелиоцитами, гладкомышечными клетками, макрофагами. PDGF является не только хемоаттрактантом, но и митогеном для гладкомышечных клеток. Далее мигрировавшие в интиму гладкомышечные клетки интенсивно пролиферируют под влиянием фактора роста фибробластов и, возможно, ФНО-α и и ИЛ-1. Фактор роста фибробластов (fibroblast growth factor, FGF) продуцируется макрофагами, гладкомышечными клетками, эндотелиоцитами. FGF стимулирует пролиферацию не только гладкомышечных клеток, но также профибробластов и эндотелиоцитов.
Сами гладкомышечные клетки секретируют ряд биологически активных веществ — цитокинов и факторов роста: ИЛ-1 (стимулирует пролиферацию Т-лимфоцитов и гладкомышечных клеток); ИЛ-6 (стимулирует пролиферацию Т- и В-лимфоцитовипревращение последнихвплазматическиеклетки);моноцитарный хемоаттрактантный протеин-1; моноцитарный колониестимулирующий фактор; фактор роста, идентичный фактору роста тромбоцитарному; трансформирующий фактор роста-β; фактор роста фибробластов.
Мигрировавшие в интиму и пролиферирующие гладкомышечные клетки приобретают новые свойства и участвуют в формировании экстрацеллюлярного соединительного матрикса будущей атеросклеротической бляшки в очаге пораже ния артериальной стенки. Экстрацеллюлярный матрикс (ЭМ) включает в себя межуточный коллаген I и III типов, протеогликаны (версикан, бигликан, аггрекан, декорин), эластин. Все эти компоненты ЭМ продуцируются гладкомышечными клетками под влиянием тромбоцитарного ростового фактора (PDGF) и трансформиру ющего ростового фактора-β (transforming growth factor-beta, TGF-β). Оба фактора продуцируются тромбоцитами, а также эндотелиальными и гладкомышечными клетками, макрофагами. Кроме того, гладкомышечные клетки приобретают способность к нерегулируемому захвату модифицированных ЛПНП путем прямого эндоцитоза, что приводит к накоплению в них эфиров холестерина. Скэвенджеррецепторов гладкомышечные клетки не имеют. Перегруженные липидами гладкомышечные клетки трансформируются в макрофагоподобные миоинтимоциты, то есть фактически превращаются в пенистые клетки миоцитарного происхождения.
В процессе атерогенеза в развивающейся атероме наряду с пролиферацией гладкомышечных клеток наблюдается их апоптоз. Последний стимулируется провоспалительными цитокинами и протекает с участием цитотоксических Т-лимфоцитов (Т-киллеров). В очаге атеросклеротического поражения наблюдается скопление цитотоксических Т-лимфоцитов. На поверхности этих лимфоцитов экспрессируется Fas-лиганд, взаимодействующий с Fas-рецептором гладкомышеч ных клеток. Fas-рецептор содержит «домен смерти», необходимый для передачи сигнала, приводящего к апоптозу. Во время гибели гладкомышечной клетки, перегруженной липидами, происходит выделение липидов в субэндотелиальное пространство интимы.
На начальных этапах атероматозного повреждения рост бляшки наблюдается преимущественно кнаружи от ламинарного потока в артериях , в то время, как рост
437
внутрь приводил бы к резкому ограничению ламинарного потока — ламинарному стенозу. Ламинарный стеноз развивается, когда растущая бляшка на 40 % уменьшает диаметр артерии. Рост интимы кнаружи от ламинарного потока приводит к увеличению калибра артерии. Это так называемое позитивное ремоделирование, или компенсаторное увеличение, сопровождается повышением синтеза ЭМ для обеспечения кругового роста артерий.
Таким образом, атеросклеротическая бляшка проходит все этапы формирования, начиная с накопления липопротеидов в интиме артерий, образования пенистых клеток,появлениялипидныхпятенилипидныхполосок,отложенияЭХисвободного ХН во внеклеточном пространстве с образованием липидного ядра и заканчивая появлением фиброзной покрышки, то есть формированием фиброатеромы.
Течение атеросклеротического поражения артерий характеризуется чередованием стабильной и нестабильной фаз. Дестабилизация атеросклеротической бляшки всегда представляет большую опасность. Основными патогенетическими факторами нестабильного течения атеросклеротической бляшки являются ее эрозия, трещины, надрывы и разрывы; тромбоз (тромбообразование начинается в месте разрыва или эрозии бляшки); сосудистый спазм; высокая активность металлопротеиназ, выделяющихся активированными макрофагами и повреждающих фибринозную оболочку бляшки ; воспаление в атеросклеротической бляшке. Истончению и разрывам соединительнотканной оболочки атеросклеротической бляшки способствуют протеолитические ферменты металлопротеиназы (коллагеназа, желатиназа, стромелизин), вырабатываемые макрофагами , тучными клетками и разрушающие экстрацеллюлярный матрикс, а также продолжающееся увеличение размеров ядра бляшки. Наличие воспаления в атеросклеротической бляшке подтверждается обнаружением в крови в значительном количестве маркеров воспалительного процесса: С-реактивного протеина, ИЛ-8. С-реактивный белок является фактором прогноза тяжелых приступов заболеваний сосудов сердца и головного мозга. Иногда атеросклеротическая бляшка становится источником эмболии в различные артерии. Осложнением атеросклеротической бляшки является также кальцификация (атерокальциноз). При кальцификации наряду с кальцием происходит отложение морфогенетических белков костной ткани — остеокальцина, остеопонтина. Наиболее часто кальцифицируются брюшная аорта, коронарные артерии, артерии таза, бедренные артерии.
Прогрессирующее развитие атеросклеротической бляшки, особенно присоединение ее осложнений, приводит к развитию критического стеноза пораженной артерии и соответственно к ишемии соответствующего органа.
17.5.6. Принципы коррекции нарушений обмена липидов и липопротеидов
1. Диета: не переходить в питании границы нормы потребления холестерина в сутки (300 мг); включать в диету ПНЖК и продукты моря, содержащие ω-3- ненасыщенные жирные кислоты (эйкозапентаеновую кислоту С 20:5 ω-3 и докозагексановую кислоту С 22:6 ω-3).
438
2.Умеренная физическая нагрузка, способствующая увеличению ЛПВП в
крови.
3.Медикаментозное лечение гиперлипопротеинемий.
Медикаментозное лечение назначают в дополнение к диете, когда она не приводит к улучшению липидного обмена. Антисклеротические препараты имеют различный механизм действия и для их рационального применения необходимо учитывать особенности их влияния на липидный обмен. Выделяют следующие основные группы антисклеротических препаратов:
1.Ингибиторы всасывания пищевого холестерина (холестирамин и аналоги).
2.Ингибиторы синтеза холестерина (клофибрат, цетамифен).
3.Препараты «ловушки» свободных радикалов и окислительномодифицированных ЛПНП (пробукол, люрсел).
4.Стимуляторы синтеза желчных кислот.
5.Ингибиторы и корректоры гипертриглицеридемий.
6.Стимуляторы обратного транспорта холестерина и синтеза ЛПВП (липостабил).
7.Ангиотропные средства, блокирующие рост или стимулирующие их частичную регрессию.
8.Средства, ускоряющие метаболизм и выведение липидов из организма:
1)препараты, содержащие ω-3-ненасыщенные жирные кислоты (ЕРА, max EPA, Elamol, Eflamol-marine);
2)гепариноиды, активирующие образование липопротеидлипазы;
9.Некоторые витамины (никотиновая кислота, пиридоксин, витамин А).
10.Липотропные средства (метионин, холина хлорид).
439
Глава 18
Патофизиология дыхания
18.1.Дыхательная недостаточность
18.1.1.Механизмы нервно-гуморальной регуляции внешнего дыхания
Как известно, различают произвольное и непроизвольное «автоматическое дыхание», причем, если автоматическое дыхание регулируется бульбарным дыхательным центром, то в произвольном или поведенчески регулируемом дыхании участвуют сигналы, исходящие из коры головного мозга, промежуточного мозга и других структур. В связи с этим очевидно, что развитие патологии дыхания может быть связано с нарушением структуры или функции в каждой из перечисленных систем, обеспечивающих дыхательный процесс. Вышеизложенное определяет целесообразность анализа современных представлений о механизмах нервногуморальной регуляции внешнего дыхания и последующих этапов транспорта кислорода к тканям.
Физиологические механизмы нервной регуляции дыхания
Дыхательный центр представляет собой совокупность нейронов продолговатого мозга, обладающих ритмической активностью и определяющих ритм дыхательных движений. Бульбарный дыхательный центр выполняет две основные функции:
1)регуляцию двигательной активности дыхательных мышц (двигательная функция);
2)гомеостатическую, связанную с изменением характера дыхания при сдвигах газового состава и кислотно-основного равновесия в крови и тканях.
Двигательная функция дыхательного центра заключается в генерации дыхательного ритма и его паттерна (длительности вдоха, выдоха, величины дыхательного объема).
Нейроны дыхательного центра расположены в дорсомедиальной и вентролатеральной областях продолговатого мозга, образуя так называемую дорсальную и вентральную дыхательные группы. В указанных дыхательных группах расположены следующие виды нейронов:
1)ранние инспираторные нейроны, максимальная частота разряда которых приходится на начало инспирации;
2)поздниеинспираторныенейроны, максимальная частота разряда — в конце инспирации;
3)полные инспираторные нейроны, характеризующиеся постоянной активностью в течение фазы вдоха;
4)постинспираторные нейроны, максимальный разряд которых обнаруживается в течение выдоха;
5)экспираторные нейроны, активность которых возрастает во второй части
выдоха;
440