

9.3.2. Строение винилбензола
В молекуле винилбензола углеводородный заместитель, представляющий систему с -связью, взаимодействует с -электронным секстетом ароматического кольца посредством, прежде всего, мезомерного эффекта: в данном случае наблюдается неполярное --сопряжение. Вследствие высокой подвижности -электронной плотности молекула винилбензола легко поляризуема и под действием катионов или полярных молекул облака электронов сильно смещаются в сторону поляризующего агента:

Следовательно, можно изобразить граничные структуры, полученные при смещении таким образом электронной плотности от винильного радикала к бензольному кольцу

Граничные структуры, полученные при обратной поляризации (например, при взаимодействии винилбензола с отрицательно заряженными ионами), значительно менее вероятны, поэтому здесь не приводятся.
Индуктивный эффект заместителя в этой молекуле практически не наблюдается, так как смещение -электронов С–С1-связи, вызванное различием электроотрицательностей атомов углерода, почти не происходит — все углеродные атомы находятся вsp2-гибридном состоянии.
Поэтому в целом, так же, как и в случае толуола, в молекуле винилбензола алкильный заместитель обладает электронодонорными свойствами по отношению к бензольному кольцу. Повышение электронной плотности происходит главным образом в о- ип-положениях к заместителю.
Геометрия винилбензола определяется валентным состоянием углеродных атомов и наличием --сопряжения. В связи с этим все атомы данной молекулы лежат в одной плоскости. Оси восьми р-орбиталей, входящих в сопряжённую систему, перпендикулярны этой плоскости. Все валентные углы близки к 120:
Однако в молекуле винилбензола, так же, как и в молекуле толуола и других замещённых бензолов, нет строгой выравненности длин и порядков связей в ароматическом кольце.
9.4. Физические свойства
Бензол и большинство алкилбензолов — жидкости, незначительно растворимые в воде, но хорошо растворимые в неполярных органических растворителях. Температура плавления в большой степени определяется симметрией молекулы. Например, п-ксилол плавится при более высокой температуре (286 К), чем его о- и м-изомеры (244 К и 220 К соответственно), что определяется более плотной упаковкой в кристалле последнего. Температуры кипения увеличиваются по мере увеличения молекулярных масс.
Алкилбензолы — малополярные соединения; причём дипольный момент симметричных соединений близок нулю, а большинства несимметричных — лежит в пределах от 0.4 до 0.6 D.
9.5. Химические Свойства
Химические свойства бензола и его алкилзамещённых должны определяться, с одной стороны, возможностью протекания реакций по ароматическому кольцу, а с другой стороны — способностью к взаимодействиям, связанным с наличием боковых углеводородных цепей.
9.5.1. Химические свойства бензола
Для бензола, обладающего устойчивой циклической -электронной системой, должны быть характерны в первую очередь реакции с электрофилами, но не реакции присоединения, как это характерно для этиленовых и полиеновых систем, а реакции замещения с сохранением устойчивой ароматической системы кольца.
Среди других реакций бензола можно отметить реакции присоединения, окисления и изомеризации. Но они менее характерны и протекают в жёстких условиях и/или с низким выходом.
9.5.1.1. Реакции электрофильного замещения
Для незамещённого бензола характерны прежде всего наиболее типичные SE-реакции: нитрование, сульфирование, галогенирование, алкилирование и ацилирование.
Механизм реакции электрофильного замещения атома водорода в бензольном кольце включает несколько стадий:
1) образование -комплекса за счёт взаимодействия -электронной системы бензола с вакантными (или частично вакантными) орбиталями электрофильной частицы

2) превращение -комплекса в карбокатион (бензолениевый или, в общем случае, аренониевый катион), который также называют -комплексом

Новая -связь С–Е образована за счёт пары электронов -системы, в результате в сопряжении с участием р-орбиталей пяти углеродных атомов задействовано только четыре электрона (энергия сопряжения составляет 109 кДж/моль); однако -комплекс не обладает ароматическим характером, а распределение электронной плотности вследствие р--сопряжения можно изобразить тремя граничными структурами:

3) разрыв -связи С–Н, возвращение пары электронов в -систему с восстановлением ароматичности; при этом может образоваться новый малоустойчивый -комплекс, который быстро депротонируется

Отщепление протона обычно протекает легко, при этом образование и разрушение промежуточного -комплекса может протекать незаметно.
Первые две стадии этой реакции аналогичны реакции электрофильного присоединения к алкенам, тогда как третья отличается от неё. Вместо присоединения аниона из реакционной среды к карбокатиону на третьей стадии наблюдается отщепление протона. Причина кроется в образовании энергетически более устойчивой ароматической системы по сравнению с диеновой:
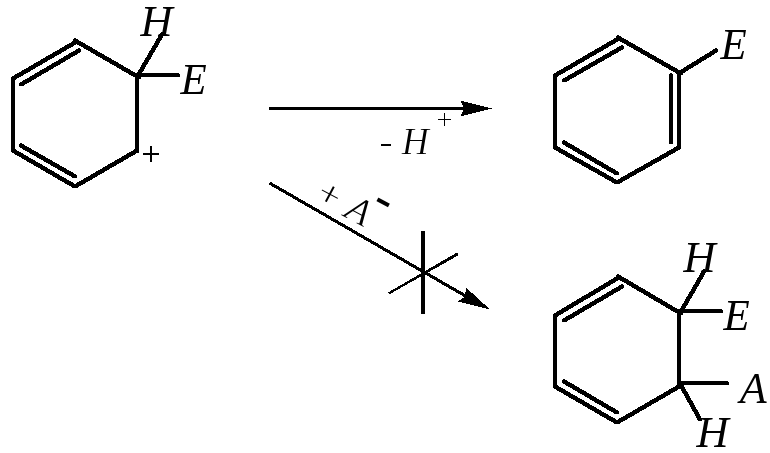
Таким образом, в большинстве случаев (реакции нитрования, галогенирования, алкилирования и ацилирования) самой медленной стадией является стадия образования -комплекса, который быстро превращается в продукт электрофильного замещения. Однако в случае сульфирования лимитирующей стадией является отщепление протона. Это можно считать одной из причин обратимости реакции сульфирования.
Рассмотрим частные случаи этих реакций.
Нитрование
Механизм реакции нитрования бензола можно представить следующим образом:
 -комплекс-комплекс
нитробензол
-комплекс-комплекс
нитробензол
Для нитрования бензола используются концентрированная азотная кислота или смеси азотной и серной кислот различных концентраций (нитрующие смеси).
Необходимая для реакции нитрования электрофильная частица, нитроний-катион NO2+, образуется по одной из возможных схем. В концентрированной азотной кислоте:
3 HNO3 NO2+ + H3O+ + 2 NO3¯
В смеси концентрированных азотной и серной кислот:
HNO3 + 2 H2SO4 NO2+ + H3O+ + 2 HSO4¯
Вместо серной кислоты могут быть использованы фтороводородная, селеновая или хлорная кислоты.
Для получения нитробензола используют нитрующую смесь в количестве, соответствующем 0.97—1.01 моля азотной кислоты на 1 моль бензола. Нитрование протекает гладко при 315—330 К. При нитровании азотной кислотой (без серной) концентрация азотной кислоты должна быть около 60%. Снижение концентрации азотной кислоты до 50% приводит к очень сильному замедлению течения реакции.
Добавление воды к нитрующей смеси и уменьшение доли серной кислоты приводит к уменьшению скорости реакции. Так, для 0.2 М раствора азотной кислоты в 100%-й серной кислоте степень превращения HNO3 в NO2+ составляет 100%, а для 0.2 М раствора азотной кислоты в 87%-й серной кислоте эта степень превращения — 14.7%. При содержании в нитрующей смеси 90% серной кислоты и 10% азотной кислоты степень превращения HNO3 в NO2+ составляет 100%, а в чистой азотной кислоте эта величина — 1.2%.
Сульфирование
Сульфирование — это реакция введения в молекулу органического соединения сульфогруппы, протекающая по схеме:

бензолсульфокислота
Электрофильными частицами реакции сульфирования могут быть катион H3SO4+ или полисерные кислоты H2S2O7, H2S3O10, H2S4O13 и т.д., хлорсульфоновая кислота ClSO3H, а также триоксид серы (серный ангидрид) SO3 и его комплексы с электронодонором (например, с диоксаном, с пиридином – гл. 3.4.4, 12.2.3.2, 12.4.3.1).
Катион H3SO4+ образуется в серной кислоте концентрации около 80% по схеме:
2 H2SO4 H3SO4+ + HSO4¯
__________________________________________________________________________________________________
В водном растворе с концентрацией серной кислоты существенно ниже 80% сульфирующая частица не образуется вследствие того, что кислота практически полностью диссоциирована:
H2SO4 + H2O H3O+ + HSO4¯,
поэтому разбавленная кислота не сульфирует.
При более высокой концентрации серной кислоты в интервале 85—98% основной сульфирующей частицей является дисерная кислота H2S2O7, которая образуется по схеме:
3 H2SO4 H2S2O7 + H3O+ + HSO4¯
__________________________________________________________________________________________________
В 100%-й серной кислоте и в олеуме (раствор SO3 в H2SO4) сульфирование происходит H2S2O7, другими полисерными кислотами и несвязанным в комплекс SO3. Сульфирование с участием SO3 протекает также при использовании его растворов в SO2 или в CH2Cl2. (Описание механизма сульфирования с участием катиона HSO3+ является достаточно условным. Этот катион можно представить как протонированную молекулу SO3 или как катионную часть дисерной кислоты H2S2O7 = [HSO3]+ [HSO4]¯.)
Механизм реакции с участием катиона H3SO4+ можно представить следующим образом:
 -комплекс -комплекс
бензолсульфокислота
-комплекс -комплекс
бензолсульфокислота
а при сульфировании дисерной кислотой —

При этом отрыв протона в последнем случае происходит либо гидросульфат-ионом HSO4¯, либо молекулой серной кислоты (в зависимости от содержания в среде воды). При сульфировании с участием SO3 механизм можно представить в следующем виде:
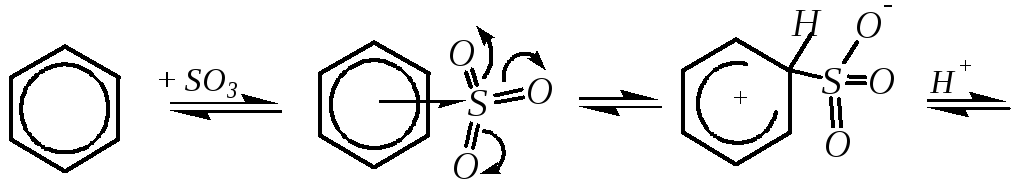

Существенным отличием реакции сульфирования от других реакций электрофильного замещения является её обратимость, хотя и некоторые другие SE-реакции могут протекать в обратном направлении. Причины обратимости сульфирования связаны с возможностью электрофильного замещения сульфогруппы на атом водорода — гл. 10.3.3.2.
Побочными реакциями в процессах сульфирования могут быть:
1) образование сульфонов (в избытке субстрата)

дифенилсульфон
2) окисление субстрата (серной кислотой или олеумом);
3) реакция десульфирования, то есть гидролиз арилсульфокислот — протекание реакции, обратной сульфированию

Галогенирование
Из всех реакций галогенирования наиболее широко используются хлорирование и бромирование. Прямое фторирование практически не используется вследствие очень высокого теплового эффекта реакции, и фторозамещенные бензолы получают косвенным путём. Йод из всех галогенов обладает самой низкой реакционной способностью в реакциях электрофильного замещения, обычно требующих присутствия в этих условиях окислителя для образования электрофильной частицы, поэтому и йодопроизводные также удобнее получать косвенным путём.
Хлорирование и бромирование ароматических соединений в ядро представляет собой типичную реакцию электрофильного замещения, но протекающую в достаточно жёстких условиях и поэтому требующую применения катализаторов. В качестве последних часто используют кислоты Льюиса, такие как галогениды железа (III), цинка и алюминия. Активность металлического железа, например, используемого в качестве катализатора, объясняется наличием в среде хлорида или бромида железа (III):
Fe + Cl2 FeCl3
Катализирующее действие кислот Льюиса может быть представлено следующей схемой:
FeCl3 + Cl2 FeCl4¯ + Cl+,
но так как хлороний-катион Cl+ не выделен, то принято считать, что образуется комплекс
Cl . . . Cl ־ . . . FeCl3 ,
который и является хлорирующим агентом. Поэтому механизм реакции можно представить в следующем виде:
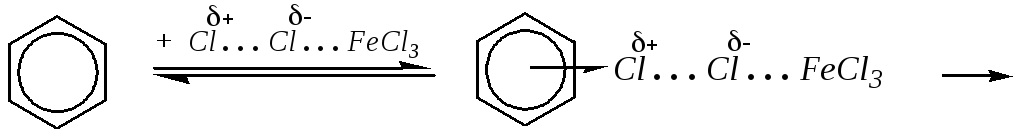
-комплекс

-комплекс хлорбензол
Хлорирование в ароматическое ядро может катализироваться не только железом, но также серной кислотой или молекулярным йодом. В этих случаях образование хлороний-катиона протекает по следующим схемам:
Cl2 + H2SO4 Cl+ + HSO4¯ + HCl
Cl2 + I2 2 ICl
ICl I+ + Cl¯
I+ + Cl2 ICl + Cl+
Однако если хлор и йод взяты в эквимолекулярном соотношении, то последняя стадия не осуществляется, хлороний-катион не образуется, но может идти йодирование за счет йодоний-катиона, образующегося на предыдущей стадии.
Ацилирование
Ацилирование — это реакция замещения атома водорода ацильным остатком (ацилом). Ацилирование, как и алкилирование (см. далее) ароматических соединений, — это реакция Фриделя–Крафтса*. Наиболее широко распространены ацетилирование, то есть введение ацетильной группы CH3CO-, и формилирование — введение формильной группы HCO-. Обычно ацилирование в бензольное кольцо протекает только с использованием галогенангидридов кислот и обязательно в присутствии катализаторов — кислот Льюиса, обычно AlCl3. Механизм с участием катализатора может быть такой:


-комплекс

-комплекс
Формилирование может проводиться смесью монооксида углерода и хлороводорода (реакция Гаттермана*–Коха):

Электрофильная частица здесь образуется по схеме:
![]()
Ацилирование аренов с помощью фосгена — прямой способ получения галогенангидридов ароматических карбоновых кислот:

Своеобразным вариантом ацилирования является реакция галогенометилирования (реакция Блана*):
 ,
,
которая может служить одним из способов получения бензилгалогенида и его производных. Это обычная реакция электрофильного замещения, в которой электрофильная частица образуется при протонировании формальдегида:
H2C=O
+ HCl
![]() [+CH2OH]
Cl¯
[+CH2OH]
Cl¯


Алкилирование
Алкилирование бензола (как и многих ароматических соединений) по Фриделю–Крафтсу осуществляется галогеналканами в присутствии кислот Льюиса. Но в общем случае алкилирование в ароматическом ряду может осуществляться также спиртами и алкенами в присутствии кислотных катализаторов.
В зависимости от природы радикала R роль катализатора в реакции может быть различной. Он может лишь способствовать образованию карбокатиона R+:
R-Cl + AlCl3 R+ + AlCl4¯
или
R-OH + H+ R+ + H2O,
являющегося электрофильной частицей и атакующего молекулу бензола. Но если образование карбокатиона невозможно (из-за его неустойчивости), то катализатор, образуя с молекулой реагента промежуточный комплекс, участвует в электрофильной атаке по бензольному кольцу. Например:
![]()

-комплекс

-комплекс
Поэтому легче реакция алкилирования протекает с такими алифатическими реагентами, которые образуют устойчивые карбокатионы. Это преимущественно реагенты аллильного типа и третичной структуры.
Однако существенное влияние может оказывать природа катализатора и природа атомов галогена в составе алкилгалогенидов. Так, например, при взаимодействии бензола с неопентилхлоридом в присутствии AlCl3 получается исключительно трет-амилбензол:
 ,
,
так как ещё до реакции с бензолом успевает пройти изомеризация первичного карбокатиона в третичный:

Однако если в качестве катализатора используется FeCl3, то основным продуктом является неопентилбензол, образующийся из неизомеризованного иона.
Следует иметь в виду, что реакцию алкилирования трудно остановить на стадии моноалкилирования, образуются ди- и полиалкилпроизводные. Это объясняется тем, что продукты моноалкилирования, алкилбензолы, вступают в эту реакцию легче, чем сам бензол (см. далее, в главе 9.5.2.1). Поэтому для получения моноалкилбензолов обычно используют избыток бензола.
В некоторых случаях реакции алкилирования могут быть обратимы. Это выражается в возможности электрофильного замещения алкильной группы на другую функциональную группу (например, при реакциях нитрования ароматических соединений). Однако такие случаи достаточно редки (гл. 10.5.3.3), причём замещаемая алкильная группа должна быть пространственно доступна, а уходящий алкильный катион должен быть достаточно стабильным.
9.5.1.2. Реакции присоединения
Гидрирование
В отличие от алкенов бензол присоединяет водород в жёстких условиях при высокой температуре (370—470 К) и давлении (3—10МПа) с использованием в качестве катализатора пористого никеля (так называемогоникеля Ренея), при этом происходит разрушение ароматической системы и исчерпывающее гидрирование всего соединения.
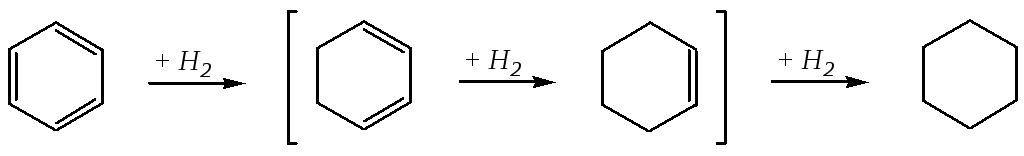
В реакции каталитического гидрирования молекулярным кислородом удаётся выделить только конечный продукт — циклогексан. Выделить промежуточные продукты (циклогексадиен, циклогексен) не удаётся, так как они гидрируются быстрее, чем бензол. Это обусловлено тем, что поглощение первого моля водорода протекает со значительно бóльшими энергетическими затратами из-за необходимости нарушения ароматического секстета, тогда как остальные два моля водорода присоединяются почти мгновенно с выделением энергии.
Однако восстановлением по Бёрчу*можно получить циклогексадиен-1,4. Это восстановление щелочными металлами (обычно натрием), растворёнными в жидком аммиаке (или амине), в присутствии спирта. Механизм реакции включает образование на первой стадии анион-радикала, который затем селективно протонируется в том месте, где электронная плотность наиболее высокая:


циклогексадиен-1,4
Присоединение хлора
Хлор присоединяется к бензолу только при интенсивном облучении светом. Присоединяются сразу три моля хлора (по той же причине, что и при гидрировании присоединяются сразу три моля водорода).

Реакция имеет радикальный механизм (АdR), при этом образуется несколько геометрических изомеров, соответствующих приведённой структуре, один из них — гексахлоран, обладающий сильными инсектицидными свойствами.
В эту реакцию не вступают производные бензола. Присоединение хлора характерно только для незамещённого бензольного кольца.
9.5.1.3. Фотохимическая изомеризация
При ультрафиолетовом облучении бензол изомеризуется, теряя ароматические свойства и превращаясь в бензвален, дьюаровский бензол, призман и фульвен. Все эти углеводороды очень неустойчивы и легко переходят в бензол.
Бензвален (или бензол Хюккеля) образуется с выходом 1% при длительном облучении жидкого бензола светом длиной волны 254нм:

бензвален
Бензол Дьюара получают при быстром облучении бензола светом длиной волны 206 нм:

бензол Дьюара
Призман (бензол Ладенбурга*) — продукт внутримолекулярного циклоприсоединения бензола Дьюара:

призман
Образование фульвена можно представить следующим образом:

фульвен
9.5.1.4. Реакции окисления
Бензол устойчив к окислителям при комнатной температуре. При действии на бензол кислорода воздуха в присутствии оксида ванадия (V) при температуре700Кпроисходит разрушение бензольного кольца с образованием малеинового ангидрида. Это промышленный способ получения малеинового ангидрида. В процессе реакции как бы окисляется диеновая система бензола:

Под действием ферментов (например, в печени кролика) бензол может окисляться в муконовую кислоту. Эта реакция, протекающая с разрывом С=С-связи, аналогична алкенам:

муконовая кислота
Озонолиз
Действие озона на бензол аналогично действию на непредельные соединения и приводит к образованию триозонида:
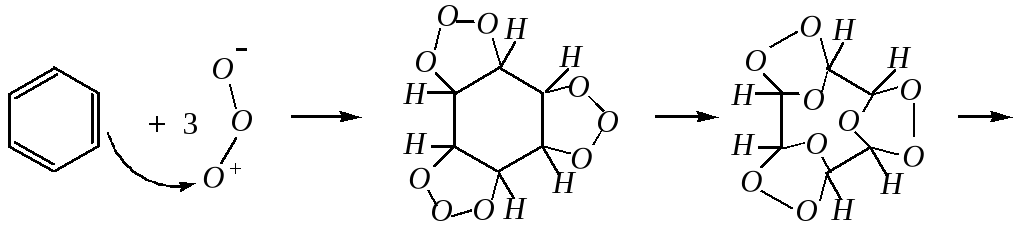

глиоксаль
Расщепление озонида с образованием глиоксаля может происходить также, если вместо гидролиза использовать восстановление молекулярным водородом в присутствии катализатора:
