

План выполнения практической работы:
1. Проведение реакций идентификации прокаина гидрохлорида.
2. Проведение количественного определения прокаина гидрохлорида нитритометрическим методом.
3.Написание отчета.
Примечание. Практическую работу выполнять по методикам ФС 42-0265-07, ГФ XII.
Указания при проведении практической работы
Каждый студент группы самостоятельно проводит идентификацию и количественное определение прокаина гидрохлорида по методикам, указанным в ФС 42-0265-07, ГФ XII(см. Приложение 1):
При составлении отчета указать:
латинское, международное непатентованное, химическое название прокаина гидрохлорида, указать его структурную формулу;
описание внешнего вида прокаина гидрохлорида;
по реакциям идентификации препарата дать теоретическое обоснование метода, написать уравнения реакций и указать результаты анализа;
по количественному определению препарата дать теоретическое обоснование метода, методику, написать уравнения химических реакций, fэкв. , м.м.э., w (%) и указать результаты анализа(сделать вывод).
Приложение 1
Новокаина гидрохлорид (ФС 42-0265-07)
[2-(Диэтиламино)этил]-4-аминобензоата гидрохлорид
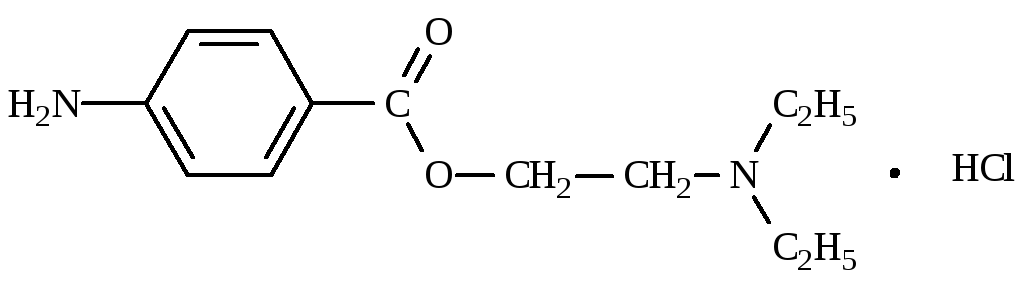
|
C13H20N2O2·HCl |
М.м. 272,78 |
Cодержит не менее 99,5 % и не более 101,0 % C13H20N2O2·HCl в пересчете на сухое вещество.
Описание. Белый кристаллический порошок или бесцветные кристаллы.
Растворимость. Очень легко растворим в воде, растворим в спирте 96 %, мало растворим в хлороформе.
Подлинность. Инфракрасный спектр субстанции, снятый в диске с калия бромидом, в области от 4000 до 400 см-1 по положению полос поглощения должен соответствовать рисунку спектра новокаина гидрохлорида (Приложение 1).
0,05 г субстанции растворяют в 2 мл воды. Раствор дает характерную реакцию на первичные ароматические амины с образованием оранжево-красного окрашивания, переходящего в вишнево-красное.
0,05 г субстанции растворяют в 2 мл воды, прибавляют 0,15 мл серной кислоты разведенной 16 % и 1 мл 0,1 М раствора калия перманганата; фиолетовое окрашивание сразу исчезает.
Субстанция дает характерную реакцию на хлориды.
Температура плавления. От 154 до 158 ºС.
Прозрачность раствора. Раствор 1 г субстанции в 10 мл воды должен быть прозрачным или выдерживать сравнение с эталоном I.
Цветность раствора. Раствор, полученный в испытании на Прозрачность раствора, должен быть бесцветным или выдерживать сравнение с эталоном В9.
рН. От 6,0 до 7,5 (1 % раствор).
Посторонние примеси. Определение проводят методом ТСХ.
Испытуемый раствор. 0,2 г субстанции растворяют в 0,6 мл воды и разбавляют спиртом 96 % до 10 мл.
Раствор сравнения. 0,01 г 4-аминобензойной кислоты и 0,01 г анестезина растворяют в 100 мл спирта 96 %. 1 мл полученного раствора разбавляют спиртом 96 % до 10 мл.
На линию старта пластинки со слоем силикагеля 60 F254 наносят 20 мкл (400 мкг) испытуемого раствора и 20 мкл (0,2 мкг 4-аминобензойзойной кислоты и 0,2 мкг анестезина) раствора сравнения. Пластинку с нанесенными пробами сушат на воздухе, помещают в камеру со смесью бензол - ацетон (4:1) и хроматографируют восходящим методом. Когда фронт подвижной фазы дойдет до конца пластинки, ее вынимают из камеры, сушат на воздухе и просматривают в УФ свете при 254 нм.
Пятна посторонних примесей на хроматограмме испытуемого раствора, находящиеся на уровне пятен 4-аминобензойной кислоты и анестезина, по совокупности величины и интенсивности поглощения не должны превышать соответствующие пятна на хроматограмме раствора сравнения (не более 0,05 % каждой примеси).
Потеря в массе при высушивании. Около 1,0 г (точная навеска) субстанции сушат при температуре от 100 до 105 ºС до постоянной массы. Потеря в массе не должна превышать 0,5 %.
Сульфатная зола и тяжелые металлы. Сульфатная зола из 1,0 г (точная навеска) субстанции не должна превышать 0,1 % и должна выдерживать испытание на тяжелые металлы (не более 0,001 % в субстанции).
Остаточные органические растворители. В соответствии с требованиями ОФС «Остаточные органические растворители».
Бактериальные эндотоксины. Не более 0,14 ЕЭ на 1 мг субстанции.
Для проведения испытания готовят исходный раствор субстанции (концентрация 100 мг/мл), а затем разводят его не менее чем в 400 раз.
Испытание проводят для субстанции, предназначенной для приготовления стерильных лекарственных форм.
Микробиологическая чистота. В соответствии с требованиями ОФС «Микробиологическая чистота».
Количественное определение. Около 0,3 г (точная навеска) субстанции растворяют в смеси 10 мл воды и 10 мл хлористоводородной кислоты разведенной 8,3 %. Полученный раствор титруют нитритометрически.
Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора натрия нитрита соответствует 27,28 мг C13H20N2O2·HCl.
Хранение. Список Б. В сухом, защищенном от света месте.
Приложение 2
НИТРИТОМЕТРИЯ (ОФС 42-0054-07)
Нитритометрия – метод титриметрического анализа, при котором в качестве реактива для титрования используется раствор натрия нитрита.
Применяется для количественного определения соединений, содержащих первичную или вторичную ароматическую аминогруппу, для определения гидразидов, а также ароматических нитросоединений после предварительного восстановления нитрогруппы до аминогруппы.
Методика. Около 0,001 грамм-моля (точная навеска) образца лекарственного средства растворяют в смеси 10 мл воды и 10 мл хлористоводородной кислоты разведенной 8,3 %. Прибавляют воду до общего объема 80 мл, 1 г калия бромида и при постоянном перемешивании титруют 0,1 М раствором натрия нитрита. В начале титрования прибавляют раствор натрия нитрита со скоростью 2 мл/мин, а в конце (за 0,5 мл до эквивалентного количества) – 0,05 мл/мин.
Титрование проводят при температуре раствора 15-20 0С, однако, в некоторых случаях требуется охлаждение до 0 – 5 0С.
Точку эквивалентности определяют электрометрическими методами (потенциометрическое титрование, амперометрическое титрование) или с помощью внутренних индикаторов.
При потенциометрическом титровании в качестве индикаторного применяют платиновый электрод, при этом в качестве электродов сравнения используют хлорсеребряный или насыщенный каломельный электрод.
На электроды накладывают разность потенциалов 0,3 – 0,4 В, если не указано иначе в частной фармакопейной статье.
В качестве внутренних индикаторов используют тропеолин 00 (4 капли раствора), тропеолин 00 в смеси с метиленовым синим (4 капли раствора тропеолина 00 и 2 капли раствора метиленового синего), нейтральный красный (2 капли в начале и 2 капли в конце титрования).
Титрование с тропеолином 00 проводят до перехода окраски от красной к желтой, со смесью тропеолина 00 с метиленовым синим – от красно-фиолетовой к голубой, с нейтральным красным – от красно-фиолето-вой к синей. Выдержку в конце титрования с нейтральным красным увеличивают до 2 мин.
Параллельно проводят контрольный опыт.