

Наиболее схожие по структуре белки
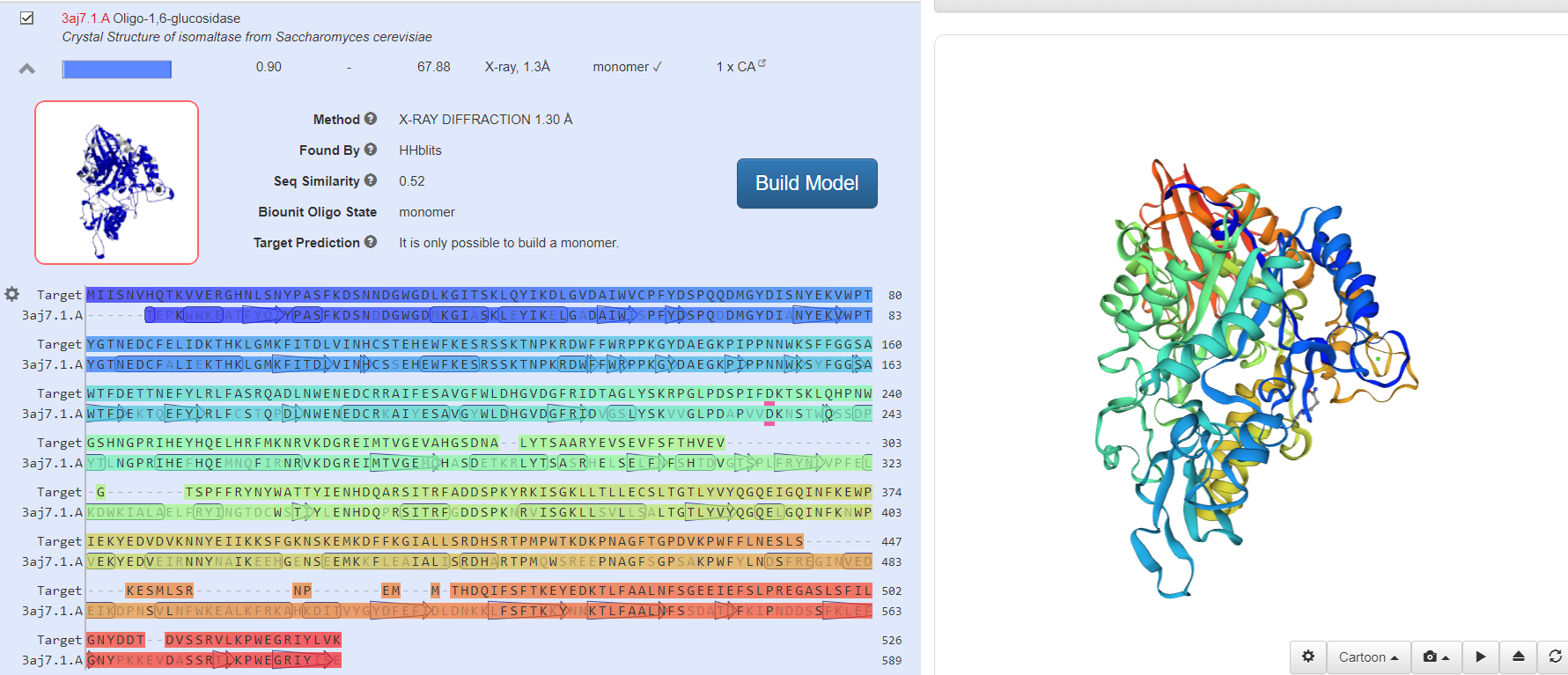
Модель выбранного белка после «Build Model»


Модель выбранного белка после «Build Model»


Модель выбранного белка после «Build Model»

Вывод: Входе лабораторной работы смоделировали третичную структуру белка, взяв первичную структуру с сайта https://www.ncbi.nlm.nih.gov. Сравнили сходство полученной модели амилазы с известными структурами других организмов. Структуры полученных белков имеют высокое сходство с изначально выбранными образцами.
Короновирус (SARS-CoV-2) проникает к клетки после контакта со специфическим белком (АСЕ2 - ангиотензинпревращающий фермент). Если на поверхности клетки его нет, то вирус не сможет ее инфицировать (подробнее на https://en.wikipedia.org/wiki/Severe_acute_respiratory_syndrome_coronavirus_2#/media/File:SARS-CoV-2_without_background.png ).
На данный момент на ресурсе https://www.expasy.org/ приведены последние актуальные данные по SARS-CoV-2, геном, протеины и др. Особо полезную информацию найдут исследователи, которые методами молекулярного моделирования (in silico) пытаются создать лекарство против вируса. Возможность найти мишени (протеины), на которые будут действовать (связываться) молекулы. В частности, если «заблокировать» АСЕ2 низкомолекулярным лигандом, то это будет препятствовать адсорбции вируса на поверхности клетки, и как следствие этого уменьшать вероятность проникновения его в клетку.
На ресурсе https://www.uniprot.org/uniprot/Q9BYF1 информация про АСЕ2 (ангиотензинпревращающий фермент). Было выяснено, что замена некоторых аминокислотных остатков в АСЕ2 предотвращает связывание с вирусом. Замена К (лизин) на D (аспарагиновая кислота) в положении 31 предотвращает взаимодействие с гликопротеином «шпионом», как и замена тирозина (Y) на аланин (A) (Обведено).

Fasta файл , первичная последовательность аминокислот
>sp|Q9BYF1-2|ACE2_HUMAN Isoform 2 of Angiotensin-converting enzyme 2 OS=Homo sapiens OX=9606 GN=ACE2
MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEENVQ
NMNNAGDKWSAFLKEQSTLAQMYPLQEIQNLTVKLQLQALQQNGSSVLSEDKSKRLNTIL
NTMSTIYSTGKVCNPDNPQECLLLEPGLNEIMANSLDYNERLWAWESWRSEVGKQLRPLY
EEYVVLKNEMARANHYEDYGDYWRGDYEVNGVDGYDYSRGQLIEDVEHTFEEIKPLYEHL
HAYVRAKLMNAYPSYISPIGCLPAHLLGDMWGRFWTNLYSLTVPFGQKPNIDVTDAMVDQ
AWDAQRIFKEAEKFFVSVGLPNMTQGFWENSMLTDPGNVQKAVCHPTAWDLGKGDFRILM
CTKVTMDDFLTAHHEMGHIQYDMAYAAQPFLLRNGANEGFHEAVGEIMSLSAATPKHLKS
IGLLSPDFQEDNETEINFLLKQALTIVGTLPFTYMLEKWRWMVFKGEIPKDQWMKKWWEM
KREIVGVVEPVPHDETYCDPASLFHVSNDYSFIRYYTRTLYQFQFQEALCQAAKHEGPLH
KCDISNSTEAGQKLL
Если найти соединения, которые будут связываться с остатками аминокислот K31, Y41, D355 и др., то можно надеяться (если не будет побочных эффектов, если фармакокинетические параметры будут приемлемые - ADMET), что получатся препараты для профилактики заражения короновирусной инфекцией.
https://www.ebi.ac.uk/pdbe/entry/pdb/1R42 структура АСЕ2


Для теоретической оценки взаимодействия белка (ACE2) с какой либо молекулой (лиганд) используется т.н. процедура докинга (дословный перевод с англ. – стыковка, причаливание) https://ru.wikipedia.org/wiki/%D0%9C%D0%BE%D0%BB%D0%B5%D0%BA%D1%83%D0%BB%D1%8F%D1%80%D0%BD%D1%8B%D0%B9_%D0%B4%D0%BE%D0%BA%D0%B8%D0%BD%D0%B3
В большинстве случаев программы для докинга платные, но существуют и бесплатные. В частности на интернет ресурсе expasy.org есть бесплатная (free of charge), общедоступная онлайн программа для молекулярного моделирования (докинга) - SwissDock.
Таким образом, алгоритмом теоретического поиска «возможного лекарства от короны» является:
1. На основании вышеизложенных данных, провести «докинг» ACE2 с какой-либо молекулой. Получить оценку силе взаимодействия и увидеть место «стыковки», т.е. с какими аминокислотными остатками АСЕ2 будет взаимодействовать тестируемая молекула и как сильно.
2. Необходимо найти на ресурсе Pdb.org графическую структуру докируемого белка (или его код) и сохранить в pdb – расширении (например, 1r42.pdb) на рабочий стол.
3. Нарисовать в химическом редакторе (chemdraw или др. доступный) структуру соединения (можно протестировать известные противовирусные препараты или придумать свою собственную молекулярную органическую структуру), и после оптимизации ее методом молекулярной механики (Chem3D) сохранить в mol2 расширении.
База данных Pdb.org содержит информацию о 3D-структуре белка, полученную рентгеноструктурным методом (X-ray) (как правило). Предварительно интересующий белок кристаллизуют из воды. Поэтому в графическом файле помимо самого белка присутствуют молекулы воды (как растворитель) и вспомогательные соединения (глицерин, ионы буферных растворов, ПАВы). Для корректного докинга их необходимо удалить. Поэтому лучше использовать уже подготовленный файл, где в соответствующей программе (все химические редакторы, которые работают с макромолекулами - белками) удалено лишнее (ACE2prep2.pdb). Этот файл загружать в SwissDock, он находится на моей странице!!!
На ресурсе expasy.org в категории proteomics (1) открыть в Tools вкладку (2) и в разделе submit docking в target selection загрузить (с помощью search найти его) с рабочего стола белок ACE2prep2.pdb и в ligand selection также тестируемую молекулу в mol2 формате и указать на английском что-нибудь в окошечке «job name», и электронный адрес, куда придут результаты. Если дождаться результатов on-line, то можно посмотреть их в интерактивной форме. Но обычно результтаты будут готовы на следующий день.
Результатом докинга будут положения лиганда в комплексе с белком и энергия взаимодействия. Чем меньше значение энергии взаимодействия, тем прочнее комплекс.




Вид результатов докинга

Результаты можно сохранить в zip.формате (Download your predictions file).

Для удобного просмотра результатов докинга необходимо скачать и установить программу Chimera. В нашем случае нам важно, чтобы не только энергия связывания (binding) была минимальная, но место связывания находилось около 31, 41 и 355 аминокислотных остатков. Поэтому визуально в JavaSmol мы этого не увидим.
Программа для визуализации результатов докинга и вообще для молекулярного моделирования Chimera для академического использования бесплатная. Ее скачать и установить можно по ссылке «You can get Chimera here» (см. )

Для просмотра результатов, необходимо скачать файл «open. chimarax». (Launch UCSF Chimera to …..) и сохранить в папке, где расположен и zip-файл.
Более полную информацию о том, как работать в «химере», можно найти в YouTube или читать раздел Help.
После установки программы открываем ее и в строке file\open находим и открываем «open. chimarax». Может всплыть предупреждение об угрозе от открытия, мы жмем Yes и тогда откроются 2 окна, с белком и результатами докинга. Для визуального анализа мест связывания необходимо выделить интересующие аминокислотные остатки. В строке Tools - sequence – sequence выделить K31 (лизин), Y41 (тирозин) и D355 (аспарагиновая кислота). Для мультивыделения использовать кнопку Shift.

Результаты докинга группируются в кластеры (что это такое?).
К
сожалению, в представленном в качестве
примера результат докирования соединения
(дикумарол)
 с
белком ACE2
ни в одном случае нет кластеров с близким
контактом лиганда с ключевыми
аминокислотными остатками (на картинке
зеленым выделены остатки лизина, тирозина
и аспарагиновой кислоты; лиганд -
голубой). Ниже приведены изображения
кластеров 21 и 22.
с
белком ACE2
ни в одном случае нет кластеров с близким
контактом лиганда с ключевыми
аминокислотными остатками (на картинке
зеленым выделены остатки лизина, тирозина
и аспарагиновой кислоты; лиганд -
голубой). Ниже приведены изображения
кластеров 21 и 22.
Цели: Найти какое-либо соединение с известными экспериментальными данными по липофильности и растворимости.


Известные данные:
Липофильность – LogP =1,2
Растворимость в воде – 3мг/мл

Различие в липофильности можно объяснить наличием -SO3H группы и йода.
Вывод: Входе лабораторной работы способна приблизительно предсказать липофильность введенного вещества на основе его структуры.
Ход работы
Условия хроматографирования:
Хроматографическая колонка: 150×3,5 мм для хроматографа «Agilent», заполненная сорбентом с размером зерен 3,5 мкм; обращенно-фазовая
Режим элюирования: градиентный;
Подвижная фаза: ацетонитрил – вода, 0 мин = 100% H2O, 20 мин = 100% CH3CN
Скорость потока элюента – 1 мл/мин;
Детектор – УФ-спектрофотометрический, λ=270 нм;
Температура колонки: 25 0С.
Объем вводимой пробы – 20 мкл.
Предварительно путем введения водных растворов соединений с известной липофильностью в хроматограф определяют время удерживания, результаты вносят в таблицу 1.
Задание.
В программе ChemDraw нарисовать структуру соединения с известной липофильностью, затем в меню View выбрать Show chemical properties window и вставили приведенное значение ln P в таблицу 1 (графа «расчетное»).
Построить график зависимости времени удерживания от липофильности.
Для аспирина (ацетилсалициловая кислота) рассчитать липофильность по экспериментально найденному времени удерживания (6.8 мин).
Для аспирина рассчитать липофильность с помощью программы ChemDraw.
Сделать выводы относительно точности предсказания липофильности расчетными методами.
Таблица 1.
Структура |
Наименование соединения |
Время удерживания (мин) |
Липофильность (lnP) |
|
Расчетное (ChemDraw) |
Экспериментальное |
|||
|
Толуол |
10,7 |
2,5 |
2,9 |
|
Бензойная кислота |
6,073 |
1,59 |
1,6 |
|
Дифениламин |
12,20 |
3,5 |
3,2 |
|
Феноксиуксусная кислота |
6,4 |
2,1 |
1,71 |
|
п-Нитроанилин |
5,65 |
1,72 |
1,38 |
|
о-Нитроанилин |
7,10 |
1,01 |
1,97 |
|
Ацетилсалициловая кислота
|
6,38 |
1,18 |
1,2 |
|
Салициловая кислота |
6,92 |
1,2 |
2,26 |









Рис.1. График зависимости времени удерживания от липофильности
Таблица 2. Сравнительные данные
Метод |
Ln(P) |
Точность расчётных данных относительно экспериментальных, % |
||
|
Ацетилсалициловая кислота |
салициловая кислота |
Ацетилсалициловая кислота |
салициловая кислота |
ChemDraw |
1,18 |
1,2 |
42 |
52 |
Эксперемен- тальное |
1,2 |
2,26 |
40 |
80,9 |
Расчётное |
1,68 |
1,828 |
|
|
Вывод: В ходе лабораторной работы рассчитали липофильность ацетилсалициловой и салициловой кислот, с помощью графика зависимости времени удержания веществ от экспериментальных значений липофильности. Расчётные значения оказались ближе к значениям полученным экспериментально.
Ход работы:
Оборудование и реактивы
Ацетилсалициловая кислота порошок;
Салициловая кислота порошок (4-нитроанилин);
Гидрохинон (4-нитрофенол) порошок
1-октанол (этилацетат)
Стеклянные баночки (пенициллинки) или делительная воронка
Плитка лабораторная электрическая
Спектрофотометр СФ-102 или аналогичный
Хроматограф Agilent Technology 1220 Infinity
Дозатор
Расчет липофильности проводят по формуле:
P= lg (Dисх.-Dконечн /(Dконечн.),
где Dконечн. – значение оптической плотности водного раствора после встряхивания с октанолом. Dисх.- оптическая плотность исходного водного раствора до встряхивания с октанолом.
Таблица 1
-
Вещество
Длина волны, нм
Оптическая плотность
4-нитроанилин
275
1,2820
Таблица 2.
Вещество |
Соотношение |
Оптическая плотность в водной фазе |
Константа распреде- ления* |
Липофильность** |
||
Водный р-р |
1-октанол (этилацетат) |
До экстракции |
После экстракции |
|||
4-нитроанилин |
4 |
1 |
1,2820
|
0,4535 |
7,307 |
0,8637 |
*
константа распределения рассчитывается
по формуле К= ** липофильность рассчитывается как логарифм константы распределения. |
||||||
 ,
где а – отношение взятого объема
водной фазы к взятому объему 1-октанола;
,
где а – отношение взятого объема
водной фазы к взятому объему 1-октанола;


Вывод: В ходе лабораторной работы было выяснено, что липофильность 4-нитроанилина составляет 0,8637, поэтому вещество является жирорастворимым.