

Факультетская_терапия_11_03_20_финал
.pdf451
больных моложе 30 лет удается индуцировать ремиссию. Критический возраст для прогноза ОМЛ — 30–60 лет. Значимость пола для прогноза меньше, чем возраст. Однако у женщин ремиссии индуцируются достоверно чаще, чем у мужчин. Неблагоприятными прогностическими факторами являются: высокая лихорадка, инфекции, кровотечения, большие экстрамедуллярные разрастания, нейролейкемия. Наличие нескольких неблагоприятных факторов приводит к резистентности к проводимой цитостатической терапии. По последним данным безрецидивные ремиссии более 5 лет наблюдаются у 30–40% у больных с ОЛЛ и 15–20% больных ОМЛ.
Говоря о перспективах научных исследований в области лечения острых лейкозов, следует назвать такие основные проблемы, как создание и изучение новых противолейкозных препаратов, в частности обладающих дифференцирующим действием на клетки костного мозга, и совершенствование методов трансплантации костного мозга.
Запомните!
Острый лейкоз — это моноклональное заболевание из группы гемобластозов, злокачественная опухоль кроветворной ткани, возникающая в результате мутации стволовой клетки крови, исходящая из костного мозга. Мутация возникает под влиянием различных мутагенных факторов и генетической предрасположенности. Патоморфологическим субстратом острых лейкозов являются лейкозные бластные клетки. Основными клиническими синдромами являются анемический, геморрагический, гиперпластический, синдром инфекционных осложнений, интоксикация. Критерием диагноза является увеличение бластных клеток в костном мозге более 20%. Принципы лечения острого лейкоза — этапность, полихимиотерапия соответственно варианту лейкоза, лечение осложнений.
Вопросы для самоподготовки
1.Определение понятия гемобластозов и острого лейкоза.
2.Этиологические факторы гемобластозов.
3.Классификация острых лейкозов (FAB).
4.Основные жалобы и варианты клинических проявлений при остром лейкозе.
5.План обследования больного с подозрением на гемобластоз.
6.Диагностические критерии острого лейкоза.
7.Дифференциальная диагностика.
8.Основные принципы и этапы лечения острого лейкоза.
9.Прогноз.
Ситуационная задача
В отделение челюстно-лицевой хирургии многопрофильного стационара поступила пациентка М. 48 лет с признаками сильного кровотечения из лунки после экстракции зуба в стоматологической поликлинике, где общий и местный гемостаз был неэффективен. За месяц до поступления отмечала явления общей слабости, субфебрилитет.
При объективном осмотре отмечаются явления стоматита, незначительное увеличение шейных и подчелюстных лимфоузлов до 1 см, безболезненных при пальпации; гепатомегалия (+2 см из-под края реберной дуги).

452
В анализе периферической крови: гемоглобин — 58 г/л, эритроциты — 1,8×1012/л, тромбоциты — 20×109/л, лейкоциты — 34×109/л, сегментоядерные — 11%, палочкоядерные — 2%, лимфоциты — 42%, моноциты — 3%, базофилы — 1%, бласты — 41%.
Вопросы:
1.Предварительный диагноз.
2.План обследования.
3.Дальнейшая тактика врача.
Литература
1.Гематология: руководство для врачей / под ред. Н. Н. Мамаева. 2-е изд., доп. и испр. СПб.: СпецЛит, 2011. 615 с.
2.Rodgers G. P., Young N. S. The Bethesda Handbook of Clinical Hematology. 2013. 512 p.
Глава 27 ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ
В общей структуре смертности от злокачественных заболеваний доля лейкозов составляет практически 10%. Заболеваемость хроническими лейкозами остается достаточно постоянной на протяжении последних десятилетий — ежегодно заболевает хроническим миелолейкозом (ХМЛ) 1 человек из 100 000. В целом количество больных хроническим миелолейкозом составляет от 15 до 20% общего числа больных лейкозами. Хронический лимфолейкоз — наиболее распространенный вид лейкоза в странах Европы, на его долю приходится около 30% всех лейкозов. Полицитемия имеет такие грозные осложнения, как нарушение микроциркуляции с ишемическим повреждением внутренних органов, тромбозы, нарушение мозгового кровообращения, инфаркт миокарда.
Хронический миелолейкоз
Хронический миелолейкоз (ХМЛ) — злокачественное миелопролиферативное заболевание из группы хронических лейкозов, хронический гранулоцитарный лейкоз.
ХМЛ характеризуется усиленным и неконтролируемым ростом миелоидных клеток с пролиферацией их до зрелых гранулоцитов (нейтрофилов, базофилов и эозинофилов). ХМЛ впервые описали в 1960 г. ученые из США — Peter Nowell и David Hungerford. ХМЛ встречается во всех возрастных группах, но чаще в среднем и пожилом возрасте. Мужчины болеют несколько чаще, чем женщины.
Этиопатогенез
Заболевание связано с мутацией ранней гемопоэтической клетки (стволовой клетки) в результате воздействия мутагенных факторов — ионизирующей радиации, химических факторов и др. В соответствии с современными представлениями ХМЛ характеризуется наличием специфической хромосомной аномалии в клетках опухолевого клона — транслокацией в 9;22 паре хромосом — филадельфийская хромосома (Ph’chromosome). Суть данной транслокации состоит в том, что часть длинного плеча 22-й хромосомы перемещается на 9-ю, а часть 9-й — на 22-ю. Данное перемещение обозначается в цитогенетике так: t(9;22). Вследствие этих перемещений 22-я хромосома значительно уменьшается в размерах и выглядит атипично. Эта хромосома и была описана как филадельфийская. Результатом транслокации является образование химерного гена bcr-abl, которым кодируется

454
специфический белок р210, обладающий тирозинкиназной активностью. Химерным данный ген назван потому, что состоит из генетического материала как 9-й, так и 22-й хромосом. Считается, что химерный ген bcr-abl и кодируемый им белок играют основную роль в патогенезе хронического миелолейкоза.
В системе гемопоэза у больных ХМЛ происходит чрезмерное неконтролируемое нарастание количества миелоидных клеток-предшественников, протекающее с вовлечением клеток гранулоцитарного, эритроидного, мегакариоцитарного и В-лимфоидного рядов. Клональная природа ХМЛ доказана путем проведения цитогенетических (обнаружение Ph-хромосомы), молекулярно-генетических (выявление химерного гена bcr-abl) и биохимических (моноклональный тип экспрессии глюкозо-6-фосфатдегидрогеназы) исследований.
Клиническая картина
Общепризнанным является выделение фаз ХМЛ: хроническая, прогрессирующая и бластная фазы ХМЛ; каждая фаза, идентифицируемая клинически, представляет собой этап в развитии лейкозного процесса. Заболевание начинается с хронической фазы, через несколько лет переходит в прогрессирующую фазу и затем — в бластный криз.
Обычно 85% пациентов ХМЛ находятся в хронической фазе на момент установления диагноза. Во время этой фазы болезнь обычно асимптоматична или малосимптомна, в этом случае заболевание выявляется случайно по анализам крови. Жалобы больных ХМЛ в хронической фазе могут ограничиваться общей слабостью, потливостью, иногда субфебрильной лихорадкой. Нередко присутствует тяжесть в левом подреберье, вызванная спленомегалией, боли в костях, суставах. Продолжительность хронической фазы варьирует и зависит от своевременной диагностики и лечения. В отсутствие лечения болезнь переходит в прогрессирующую фазу.
В настоящее время используются критерии ВОЗ для прогрессирующей (accelerated) фазы:
1)нарастание спленомегалии;
2)нарастание лейкоцитоза;
3)дополнительные цитогенетические аномалии;
4)тромбоцитопения <100×109/л;
5)базофилы в крови и костном мозге ≥20%;
6)миелобласты в крови и костном мозге 10–19%.
Переход заболевания в фазу бластного криза знаменует собой дальнейшее озлокачествление опухолевого процесса, появление новых мутаций в гемопоэтической ткани и образование в костном мозге нового опухолевого клона, характеризующегося активной пролиферацией и блокадой дифференцировки и созревания клеток. Бластный криз — финальная стадия ХМЛ и клинически протекает как острый лейкоз с быстрой прогрессией и короткой продолжительностью жизни.
Критерии бластного криза: бласты в периферической крови и в костном мозге ≥20%, экстрамедуллярные инфильтраты, состоящие из бластных клеток.
Клинический анализ крови и миелограмма. Лейкоцитоз является одним из важнейших лабораторных признаков заболевания, однако степень выраженности его
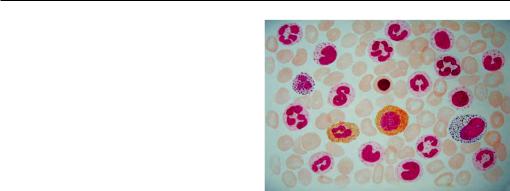

456
«излечению» заболевания. Варианты лечения ХМЛ: прицельная (направленная, векторная) лекарственная терапия (ингибиторы тирозинкиназы — дазатиниб и иматиниб), химиотерапия, биологическая терапия (интерфероны), аллогенная трансплантация стволовых клеток (ТСК).
Выполнение трансплантации аллогенного костного мозга (ТалКМ) обсуждается у всех впервые выявленных больных ХМЛ, прежде всего в начальную хроническую фазу.
Однако в связи с посттрансплантационными осложнениями в настоящее время ТалКМ используется реже.
Направленная (векторная) лекарственная терапия в настоящее время считается самой эффективной, направленной конкретно на подавление тирозинкиназы, которая содержится только в опухолевых клетках. Иматиниб (Гливек) представляет собой ингибитор тирозинкиназы, который используется для лечения первой линии во многих странах, одобрен FDA для лечения ХМЛ в 2001 г. Позже были синтезированы дазатиниб (Спрайсел) (одобрен в 2007 г.), нилотиниб, показанные в качестве препаратов второй линии при лечении больных ХМЛ в случае резистентности к предыдущему лечению или его непереносимости. Эта группа препаратов обеспечивает пятилетнюю выживаемость у 89% пациентов.
Как биологическая терапия используются препараты α-интерферона (ИФН) (роферон-А, реаферон, интрон-А, веллферон) в сочетании с цитарабином или без него. Эти препараты более эффективны в ранней хронической фазе ХМЛ. Механизм действия интерферонов-α при опухолевых заболеваниях в настоящее время окончательно не изучен, однако клинический эффект ИФН у больных с онкогематологическими заболеваниями связывают с прямым антипролиферативным действием на опухолевые клетки вследствие ингибиции экспрессии онкогенов, увеличения экспрессии мембранных антигенов опухолевыми клетками и рецепторов к гормонам; с активированием макрофагов, природных киллеров и цитотоксических эффекторных клеток; с усилением регуляторного воздействия костномозгового микроокружения: интерферон-α восстанавливает нормальное ингибирующее действие стромальных клеток на пролиферацию клеток-предшественников миелоидного ряда и усиливает адгезию последних к стромальным клеткам; с индук цией дифференцировки гемопоэтических клеток.
Интерфероны способны вызывать не только клинико-гематологическую, но и цитогенетическую ремиссию заболевания. При длительном использовании препаратов интерферона-α для лечения больных в хронической фазе ХМЛ у 46–81% пациентов наблюдается полная клинико-гематологическая ремиссия, цитогенетический ответ регистрируется у 18–56%; причем отчетливый цитогенетический ответ у 10–44% больных. Последние годы рекомендована эффективная комбинация интерферона, цитарабина и иматиниба.
Химиотерапия традиционно продолжает занимать одно из важных мест в лечении ХМЛ. При этом, как правило, используются два препарата: гидроксимочевина (гидреа, гидроксикарбамид) и миелосан (бусульфан, милеран). Эти препараты эффективны у абсолютного большинства пациентов, не являются дорогостоящими и обеспечивают высокий уровень качества жизни на этапе терапии в хронической фазе.

457
В ряде рандомизированных исследований было доказано преимущество гидроксимочевины перед миелосаном (бусульфаном): показатель выживаемости оказался выше у пациентов, принимавших гидроксимочевину в хронической фазе ХМЛ. Гидроксимочевина (гидроксикарбамид) является противоопухолевым препаратом алкилирующего действия. Цитостатический эффект достигается за счет торможения синтеза ДНК, при этом не оказывается влияния на синтез белка и РНК. Препарат применяется в дозе 2–3 г в сутки (40–50 мг/кг массы тела) в период индукции клинико-гематологической ремиссии и в дальнейшем в дозе, достаточной для поддержания ремиссии (как правило — 0,5–2 г в сутки или 10–20 мг/кг массы тела). Для оценки эффективности гидроксимочевины достаточным считается срок в 1,5 мес. Лечение проводится неограниченное время под контролем состава периферической крови.
Несмотря на очевидные преимущества гидроксимочевины перед миелосаном (бусульфаном), последний сохраняет свое место в ряду цитостатических средств, предназначенных для лечения ХМЛ. Миелосан относится к производным дисуль фоновых кислот (1,4-бис-метилсульфоновый эфир бутандиола) и действует на ранние пролиферирующие предшественники гемопоэза (преимущественно на гемопоэтические стволовые клетки) в S-фазе (фазе синтеза ДНК). Назначение миелосана оправдано у пациентов, которым не может быть проведено лечение α-интерферонами или гидроксимочевиной вследствие выраженных побочных эффектов или по другим причинам. Доза миелосана определяется количеством лейкоцитов: при лейкоцитозе 100–150×109/л назначается 8 мг миелосана, при 90–100×109/л — 6–8 мг, при 60–80×109/л — 6 мг, при 30–50×109/л — 4 мг, при 15–20×109/л — 2 мг (приведены суточные дозы). При уровне лейкоцитов ниже 15×109/л препарат назначается по 2 мг 2–3 раза в неделю. При достижении ремиссии может проводиться поддерживающее лечение — прием препарата по 2–4 мг 1–3 раза в 7–10 дней. Курсовая доза обычно составляет 250–300 мг. Следует учитывать тот факт, что миелосан действует на уровне ранних клеток-предшественни- ков гемопоэза, поэтому эффект препарата является отсроченным и в полной мере наблюдается лишь через 3–4 нед от начала терапии. Достаточно типичен подъем лейкоцитов через 10–12 дней после начала применения миелосана с последующим снижением их числа. При передозировке миелосана возможны аплазии кроветворения иногда необратимого характера, поэтому необходимо соблюдение особой осторожности при его назначении. К побочным эффектам, возникающим вследствие применения миелосана, относятся аменорея, наступающая из-за угнетения эндокринной функции яичников, пневмофиброз, причиной появления которого является повреждение альвеолярного эпителия, а также гиперпигментация кожного покрова, которая, возможно, является следствием гипокортицизма.
Интерес представляет использование малых доз цитозин-арабинозида (Аrа-С) как резервного метода лечения у пациентов в хронической фазе ХМЛ, резистентных к терапии α-интерферонами и гидроксимочевиной как в качестве средства монотерапии, так и в сочетании с этими препаратами, а также у тех больных в прогрессирующей фазе заболевания, у которых невозможно или нецелесообразно проведение полихимиотерапии. Ara-C вводится в дозе 10–15 мг/м2 2 раза в сутки подкожно в течение 10 дней ежемесячно на протяжение 4–6 мес или же в течение

458
20 дней (как правило, 2 курса с 20-дневным перерывом, в дальнейшем 20-дневные курсы повторяются по показаниям).
При наступлении бластного криза ХМЛ терапевтическая тактика вырабатывается после определения иммуноцитохимического варианта бластного криза. Актуальным остается положение о том, что лечение бластного криза ХМЛ проводится по программам, применяемым при лечении острых лейкозов. Программы полихимиотерапии часто включают в себя противоопухолевые антибиотики из группы антрациклинов (рубомицин, доксорубицин, эпирубицин, идарубицин) и цитозинарабинозид, который в настоящее время присутствует в виде двух равноценных форм (цитозар или алексан). Наиболее часто используются традиционные комбинации антрациклиновых антибиотиков и цитозин-арабинозида, такие как «5+2» и «7+3». Если удается определить иммунологический или цитохимический фенотип бластных элементов (что не всегда возможно в прогрессирующей фазе из-за небольшого количества бластных клеток) и выявляется лимфоидная их направленность, оптимальными являются комбинации препаратов, содержащих винкристин и преднизолон, такие как восьмидневный курс VRP — винкристин, доксорубицин, преднизолон, COAP — циклофосфан, онковин, адриабластин, преднизолон.
В целом переход заболевания в фазу бластного криза сочетается с крайне низкими показателями выживаемости, как правило, не превышающими 6 мес у больных с миелоидными и недифференцированными вариантами и 1 года у пациентов с лимфоидными вариантами криза.
Запомните!
ХМЛ — злокачественное миелопролиферативное заболевание из группы хронических лейкозов, хронический гранулоцитарный лейкоз. ХМЛ характеризуется усиленным и неконтролируемым ростом миелоидных клеток с пролиферацией до зрелых гранулоцитов (нейтрофилов, базофилов и эозинофилов) и их предшественников. ХМЛ характеризуется наличием специфической хромосомной аномалии в клетках опухолевого клона — транслокацией в 9;22 паре хромосом — филадельфийская хромосома. Различают хроническую, прогрессирующую и бластную фазы ХМЛ. Клинический анализ крови характеризуется гранулоцитарным лейкоцитозом с увеличением нейтрофилов, базофильно-эозинофиль- ной ассоциацией, присутствием ранних клеток миелоидного ростка — промиелоцитов, метамиелоцитов, миелоцитов, единичных миелобластов. Данные изменения сопровож даются анемией и тромбоцитопенией. Для ХМЛ характерна выраженная спленомегалия, интоксикация, субфебрилитет, анемический и геморрагический синдром. Варианты лечения ХМЛ: прицельная или векторная лекарственная терапия (ингибиторы тирозинкиназы — дазатиниб и иматиниб), химиотерапия (гидроксимочевина, миелосан), биологическая терапия (интерфероны), аллогенная трансплантация стволовых клеток (ТСК).
Ситуационная задача
Пациентка 56 лет поступила в терапевтическое отделение стационара по «скорой помощи» с диагнозом пневмонии. Рентгенологическое исследование грудной клетки подтвердило диагноз.
В клиническом анализе крови: гемоглобин — 86 г/л, эритроциты — 3,2×1012/л, тромбоциты — 68×109/л, лейкоциты — 86×109/л, сегментоядерные — 14%, палочкоядерные — 2%, базофилы — 8%, эозинофилы — 10%, миелоциты — 7%, промиелоциты — 4%, миелобласты — 1%, лимфоциты — 45%, моноциты — 9%, СОЭ — 32 мм/ч.

459
Вопросы:
1.Предварительный диагноз.
2.План обследования.
3.Дальнейшая тактика и лечение.
Хронические лейкозы лимфоидной природы
Хронический лимфолейкоз (ХЛЛ) — злокачественное опухолевое заболевание лимфатической ткани с обязательным поражением костного мозга, возникающее в результате мутации в геноме лимфоцита, при котором субстрат опухоли представлен зрелыми лимфоцитами. В соответствии с классификацией ВОЗ ХЛЛ рассматривается как лейкемическая лимфоцитарная лимфома, болезнь опухолевых В-клеток. Т-клеточный ХЛЛ сейчас называется Т-клеточной пролимфоцитарной лимфомой.
Этиологические факторы
Как самостоятельное заболевание ХЛЛ выделен в 1856 г. знаменитым немецким ученым Вирховым.
Возникновение заболевания, как и других гемобластозов, связывают с мута цией гемопоэтической (стволовой) клетки в результате воздействия ряда онкогенных факторов (ионизирующая радиация, химические и лекарственные субстанции, вирусы).
ХЛЛ преимущественно встречается у лиц пожилого и среднего возраста (средний возраст заболевших составляет 65–69 лет). Мужчины заболевают ХЛЛ в 2 раза чаще женщин. Описана наследственно-семейная предрасположенность к возникновению ХЛЛ; достаточно часто заболевание встречается у евреев.
Поскольку у больных ХЛЛ поражается лимфатическая система, играющая важную роль в иммунных реакциях организма, для этого заболевания характерны нарушения иммунитета, которые прояв ляются в склонности к заболеваниям инфекционной природы (бактериальной, вирусной и грибковой этиологии) и в предрасположенности к аутоиммунным осложнениям, самыми частыми из которых являются аутоиммунная гемолитическая анемия и аутоиммунная тромбоцитопения. Кроме того, у этих пациентов нередко обнаруживается «вторая опухоль», то есть возникающие на фоне ХЛЛ злокачественные новообразования: как правило, рак желудка, кишечника, легких. Это происходит вследствие снижения противоопухолевого иммунитета.
Клинические и лабораторные признаки ХЛЛ зависят от стадии заболевания и особенностей клинического течения.
Классификация
Классификация стадий ХЛЛ (Rai, 1975)
0 стадия — только абсолютный лимфоцитоз в периферической крови и/или костном мозге (более 5×109/л).
I стадия — абсолютный лимфоцитоз и увеличение лимфоузлов.
II стадия — абсолютный лимфоцитоз и увеличение размеров печени и/или селезенки (увеличение размеров лимфоузлов может отсутствовать).

460
III стадия — абсолютный лимфоцитоз в периферическй крови и/или костном мозге и анемия (гемоглобин менее 100 г/л).
IV стадия — абсолютный лимфоцитоз в периферической крови и/или костном мозге и тромбоцитопения (число тромбоцитов менее 100×109/л).
Классификация по BINET
Стадия А — лимфоцитоз без анемии и тромбоцитопении, лимфоаденопатия менее чем 3-х регионов (соответствует 0, 1, 2 стадии по Rai).
Стадия В — лимфоцитоз без анемии и тромбоцитопении, лимфоаденопатия более чем 3-х регионов (соответствует 1, 2 стадии по Rai).
Стадия С — лимфоцитоз с анемией и тромбоцитопенией, лимфоаденопатия (соответствует 3–4 стадии по Rai).
По рекомендациям международной группы по ХЛЛ рекомендовано применять объединенную классификацию по Rai и BINET: А(0), А(1), А(2), В(1), В(2), С(3), С(4).
Кроме того, ХЛЛ может быть разделен по особенностям клинического течения на следующие формы:
•с преобладающим поражением костного мозга;
•с преобладающим поражением лимфатических узлов;
•с преобладающим поражением селезенки;
•с выраженными аутоиммунными осложнениями (анемия, тромбоцито пения).
Клиническая картина
Жалобы пациентов в начальных (0, I) стадиях ХЛЛ могут отсутствовать или ограничиваться повышенной слабостью, иногда — потливостью, и больные в течение длительного времени (часто — несколько лет) чувствуют себя хорошо. Заболевание часто выявляется случайно при обращении к врачу по другому поводу. В большинстве случаев при осмотре удается обнаружить увеличение лимфоузлов. Лимфоузлы имеют «тестоватую» консистенцию, мягкие, подвижные, не спаянные между собой и тканями, размеры лимфоузлов могут варьировать от 1,5–2 до 10– 15 см в диаметре (поражаются подмышечные, паховые, подчелюстные, шейные и др.). Спленомегалия обычно появляется позже.
Прогрессирование заболевания связывается с возникновением и усилением общих симптомов — слабость, потливость, похудание, повышение температуры, увеличение размеров лимфоузлов. Признаки прогрессирования ХЛЛ: нарастание лимфоцитоза более чем в 2 раза за 2 мес или на 100% менее чем за 6 мес, анемия, тромбоцитопения, быстро растущие лимфоузлы, спленомегалия с селезенкой, выступающей более чем на 6 см из-под реберной дуги, потеря в весе на 10% за 6 мес, повышение температуры тела выше 38° в течение более 2 нед без признаков инфекции, ночные поты более 1 мес. Возможно развитие аутоиммунной гемолитической анемии и тромбоцитопении. Часты инфекционные осложнения (к 7–8 годам болезни — у 70% больных). Инфекции становятся причиной смерти большинства пациентов. В связи со снижением иммунитета возможно развитие злокачественных опухолей — рак желудка, рак кожи и легких.