

Физикальное обследование
Общий осмотр подразумевает оценку общего физического состояния, роста и массы тела. Выявляется бледность кожных покровов, слизистых, при тяжелой анемии присутствует учащенное сердцебиение, одышка.
Поскольку при фолиеводефицитной анемии в костном мозге происходит неэффективный эритропоэз и за счет этого повышенное разрушение гемоглобинсодержащих клеток, в крови повышается уровень непрямого билирубина и появляется легкая желтушнось склер и кожи. При тяжелых формах может наблюдаться небольшое увеличение селезенки (реактивная гиперплазия). Таким образом, имеется некоторое сходство симптоматики фолиеводефицитной анемии и гемолитической анемии. Лабораторное обследование позволяет провести надежную дифференциальную диагностику этих форм анемии.
Лабораторная диагностика
По лабораторным признакам фолиеводефицитная анемия почти совпадает с В12-дефицитной анемией. Различия заключаются в разном уровне витамина В12 и фолиевой кислоты в крови. Кроме того, для фолиеводефицитной анемии в отличие от В12-дефицитной анемии, не характерны признаки поражения нервной системы.
Для фолиеводефицитной анемии характерны:
гиперхромия, макроцитоз;
низкий уровень ретикулоцитов;
высокий уровень сывороточного железа;
высокий уровень ферритина;
умеренное повышение непрямого билирубина;
низкий уровень фолата в сыворотке крови (норма более 3 нг/мл);
низкий уровень фолата в эритроцитах;
нормальный уровень витамина В12 в крови;
картина мегалобластического кроветворения в костном мозге.
Лечение:
Детям первого года жизни фолиевая кислота назначается в дозе 0,25-0,5 мг/кг в сутки в течение месяца. В более старшем возрасте суточная доза составляет 1 мг/сутки. Пероральный прием эффективен даже в случае мальабсорбции, однако в этом случае дозу следует увеличить до 5-15 мг/сутки.
Рекомендовано в процессе лечения проводить контроль показателей крови:
Общий анализ крови с подсчетом ретикулоцитов, тромбоцитов, гематокрита - на 7 - 10 день от начала лечения. Наличие ретикулоцитарной реакции является важным признаком правильности лечения.
Контроль показателей крови 1 раз в неделю. Нормализация уровня Hb происходит через 4 - 6 недель от начала лечения и является решающим подтверждением правильности диагноза и лечения
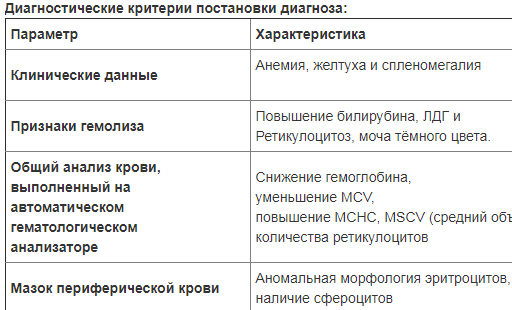
Показатель |
ЖДА |
Гемолитическая |
Гипопластическая |
В12 |
Hb |
↓ |
↓↓ |
↓↓ |
↓↓ |
Er |
↓ |
↓ |
↓↓ |
↓↓ |
ЦП |
↓ |
N |
N |
↑ |
MCV(ср V эр) |
n/↓ |
↑ |
N |
↑↑↑ |
MCH (Hb в эр) |
↓ |
↑ |
N |
↑↑↑ |
MCHC |
↓ |
N |
N |
↑ |
Pt |
N |
↑↑↑ |
↓↓↓ |
↓ |
12.Гемолитические анемии у детей – эпидемиология, этиология, классификация гемолитических анемий у детей и подростков. Патогенез анемического синдрома при гемолизе. Клиника, лабораторноинструментальная диагностика. Дифференциальная диагностика гемолитических анемий. Основные принципы лечения гемолитических анемий. Осложнения. Исходы. Профилактика. Особенности диспансерного наблюдения.
Гемолитические анемии – это группа заболеваний, характеризующихся патологически интенсивным разрушением эритроцитов, повышенным образованием продуктов их распада, а также реактивным усилением эритропоэза. В настоящее время все гемолитические анемии принято делить на две основные группы: наследственные и приобретенные.
Наследственные гемолитические анемии в зависимости от этиологии и патогенеза подразделяются на:
I. Мембранопатии эритроцитов:
II. Энзимопатии эритроцитов, обусловленные дефицитом:
III. Гемоглобинопатии:
Приобретенные гемолитические анемии:
I. Иммуногемолитические анемии:
а) аутоиммунные;
б) гетероиммунные;
в) изоиммунные;
г) трансиммунные.
II. Приобретенные мембранопатии:
а) пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы – Микели);
б) шпороклеточная анемия.
III. Анемии, связанные с механическим повреждением эритроцитов:
а) маршевая гемоглобинурия;
б) возникающие при протезировании сосудов или клапанов сердца;
в) болезнь Мошкович (микроангиопатическая гемолитическая анемия).
IV. Токсические гемолитические анемии различной этиологии.
Основными синдромами при гемолитических анемиях являются:
анемия носит нормохромный, нормоцитарный, гиперрегенираторный характер с быстро нарастающей слабостью и плохой адаптацией даже к умеренному снижению гемоглобина;
желтуха, характеризующаяся желтым окрашиванием склер, слизистой оболочки полости рта, кожи;
спленомегалия различной степени выраженности.
Немедикаментозное лечение:
Режим: общий.
Диета: стол №5.
Медикаментозное лечение:
Подходы к лечению гемолитических анемий определяются формой заболевания, первостепенной задачей является устранение гемолизирующего фактора.
План мероприятий:
1) гемотрансфузия отмытых эритроцитов (при случае снижения концентрации красных кровяных телец до критических показателей);
2) для устранения симптомов (иктеричность кожи и видимых слизистых оболочек, анемия, острый болевой криз);
3) и нормализации размеров печени и селезёнки показано применять глюкокортикоидные гормоны.
4) при аутоиммунной гемолитической анемии план лечения дополняется цитостатиками, также возможно оперативный метод лечения спленэктомия.
5) При дефиците фолатов (в гемограмме нарастает показатель MCV) назначение препаратов, содержащих цианокобаламин и фолиевую кислоту [4] (фолиевая кислота при длительной гормональной терапии).
Условие передачи пациента по месту жительства:
·--педиатр (детский гематолог) по месту жительства руководствуется рекомендациями, данными специалистами стационара;
-- частота осмотра пациента с ГА составляет 1 раз в 2-4 недели в первые 3 месяца лечения, далее в зависимости от клинического состояния и гематологической динамики, но не реже 1 раза в 2 месяца.
Прогноз при гемолитических анемиях у детей зависит от этиологического фактора. Приобретенные формы и большинство наследственных имеют благоприятный исход при своевременно начатом лечении и соблюдении мер профилактики. В качестве системы питания используется диета №5 по Певзнеру, предназначенная для снижения нагрузки на билиарный тракт. Необходим охранительный режим: избегание перегрева и переохлаждений; исключение контактов с инфекционными больными; ознакомление со списком препаратов и веществ, которые могут вызвать гемолиз (хинин, сульфаниламиды, левомицетин, аспирин, нитрофураны и прочие); определение показаний и строгих противопоказаний к вакцинации (при дефиците глюкозо-6фосватдегидрогеназы, аутоиммунных анемиях иммунизация запрещена, при сфероцитозе – рекомендована).
13.Апластические анемии у детей – эпидемиология, этиология, классификация апластических анемий у детей и подростков. Патогенез анемического синдрома при аплазии. Клиника, лабораторно-инструментальная диагностика. Дифференциальная диагностика апластических анемий. Неотложная помощь при анемическом синдроме у детей и подростков, сопровождающихся аплазией. Основные принципы лечения апластических анемий. Осложнения. Исходы. Профилактика. Особенности диспансерного наблюдения.
Апластическая анемия — заболевание системы крови, характеризующееся глубокой панцитопенией, развивающейся в результате угнетения костномозгового кроветворения.
Клиническая классификация делит все виды апластической анемии на наследственные и приобретенные.
К наследственным относят анемии с полным поражением кроветворения, имеющие 2 подвида:
Фанкони — в сочетании с врожденными пороками развития;
Эстрена-Дамешека — без пороков.
А также анемию с частичным или избирательным поражением только эритроцитарного ростка (Даймонда-Блекфена)
Макроцитарная.
Приобретенная апластическая анемия (гипопластическая) включает случаи:
острого, подострого и хронического угнетения общего производства клеток крови;
с поражением только эритроцитов — парциальную, красноклеточную анемию.
Этиология устанавливается лишь у 50%
Наиболее изученными оказались врожденные формы: при анемии Фанкони отмечена четкая связь с изменениями в парных хромосомах №1 и №7. В случаях анемии Даймонда-Блекфена обнаружены мутации генов № 1, 16, 19 и 13. Возможными активаторами считают воздействие свободных радикалов-окислителей.
Другие причины делят на наружные и внутренние.
Экзогенные (наружные) включают:
Химические агенты — производные бензола, ртути, нефтепродукты.
Физическое воздействие проникающей радиации.
Лекарственные препараты — противотуберкулезные средства (Изониазид, ПАСК), Анальгин, цитостатики, сульфаниламиды, некоторые антибиотики (Стрептомицин, Тетрациклин, Левомицетин).
Инфекция — в ряде случаев доказана связь с перенесенными инфекционными заболеваниями (гриппом, ангиной, мононуклеозом), подавляющим действием на клетки крови обладают вирусы гепатита С, герпеса, Эпштейна-Барра, цитомегаловирус.
К внутренним причинам относятся:
эндокринные нарушения — выявлена связь со сниженной функцией щитовидной железы, кистозными изменениями яичников у женщин;
иммунные сдвиги — в связи с потерей в пожилом возрасте регулирующей роли тимуса (вилочковой железы).
В основе патогенеза апластической анемии лежит внутренний дефект кроветворной стволовой клетки, приводящий к нарушению ее пролиферации и дифференцировки.
Клиника:
При анемии Фанкони у ребенка обнаруживаются врожденные дефекты в костной системе (нет первого пальца на кисти руки, искривлены или отсутствуют лучевые кости). К порокам добавляются аномалии сердца и почек, маленькие глазные яблоки.
Апластическая анемия у детей начинает проявляться с четырех лет, реже в раннем возрасте. Ребенок жалуется на головные боли, усталость. Подвержен частым простудным инфекциям, носовым кровотечениям. При обследовании выявляют характерную картину крови. Болезнь принимает хроническое течение с периодами обострений.
При анемии Эстрена-Дамешека наблюдается только патология крови. Случаи очень редкие.
Анемия Даймонда-Блекфена поражает исключительно эритроцитарный росток крови. Реже наблюдались изменения костей скелета и глаз. Кровоточивость отсутствует. Кожные покровы бледные с сероватым оттенком. Рано увеличивается селезенка и печень. В анализе крови уровень тромбоцитов и лейкоцитов снижается только при значительном поражении селезенки. Хроническое тяжелое течение болезни не позволяет дожить до 20-ти лет.
Симптомы апластической анемии проявляются в периоды обострения, а болезнь приобретает прогрессирующее медленное течение. Все признаки можно подразделить на основные синдромы, в зависимости от угнетения конкретного ростка клеток крови.
Анемия — характеризуется выраженной слабостью, жалобами на головокружение, шумом в ушах, приступами сердцебиения, одышкой.
Геморрагические проявления — на коже видны синяки, не связанные с травмированием, десны рыхлые и кровоточат. Пациентов беспокоят частые носовые кровотечения. В тяжелых случаях возможно кровоизлияние в мозг.
Сниженный уровень гранулоцитов вызывает падение защитных иммунных механизмов. Пациенты часто заражаются инфекционными болезнями. Любые ранения у них осложняются присоединением нагноения окружающих тканей.
Диагноз АА устанавливается на основании клинических проявлений и данных лабораторного обследования (А-В).
• Трехростковая цитопения: анемия (гемоглобин < 110 г/л), гранулоцитопения (гранулоциты < 2,0 : 109 /л), тромбоцитопения (тромбоциты < 100,0 : 109 /л).
• Снижение клеточности костного мозга и отсутствие мегакариоцитов по данным пунктата костного мозга.
• Аплазия костного мозга в биоптате подвздошной кости (преобладание жирового костного мозга).
Диф. диагноз :
Лейкозы — чаще обнаруживается увеличение селезенки, в костном мозге много бластных клеток предшественников лейкоцитов.
Агранулоцитозы — не вызывают анемию и снижение тромбоцитов.
Болезни с увеличением печени и селезенки — гепатиты, цирроз, тромбофлебит селезеночной вены — выявляется желтушность кожи и склер, нарушение печеночных проб.
Лечение: Впервые выявленные случаи обязательно лечатся в гематологическом отделении стационарно. Только специализированная терапия позволяет подобрать нужную дозировку и оптимальный препарат.
Основные методики:
переливание донорской крови или отдельных элементов с замещающей целью;
пересадка костного мозга;
лекарства, активизирующие кроветворение.
При аутоиммунной природе переливать нельзя. Трансплантация костного мозга является самым результативным способом лечения. Перед процедурой прекращают переливания крови, чтобы снизить возможность отторжения.
В качестве иммунодепрессантов используют: Циклоспорин, антимоноцитарный и антилимфоцитарный глобулины. Комплексным препаратом этой группы является Атгам (содержит необходимые антиглобулины). Он показан в случаях невозможности пересадки костного мозга. Для предупреждения анафилактических реакций применяют кортикостероиды.
Стимуляцию кроветворения в костном мозге осуществляют применением таких препаратов, как Филграстим, Лейкомакс. Они активизируют выработку гранулоцитов, поэтому показаны только при лейкопении. Курс лечения – две недели.
Доказана способность мужских половых гормонов (андрогенов) стимулировать все ростки крови. Для лечения мужчин применяются длительные курсы Тестостерона пропионата, Сустанона.
Удаление селезенки дает эффект у 85% больных. Метод основан на механизме прекращения выработки антител на собственные клетки. Можно проводить всем пациентам, не имеющим инфекционных осложнений
Если у больного имеется кровоточивость, вводятся кровоостанавливающие средства: Дицинон, Аминокапроновая кислота.
На сегодняшний день так и не найдено универсальное средство лечения, поэтому прогноз для жизни пациента остается неблагоприятным.
Наиболее высокая смертность в группе больных с тяжелой формой болезни. Восстановить кроветворение не удается, а больные погибают от общего сепсиса.
При менее тяжелом течении и хорошей реакции на пересадку стволовых клеток и иммунодепрессанты положительные результаты получают от половины до 90% случаев.
Условия передачи пациента педиатру (гематологу) по месту жительства.
·пациент передается под наблюдение по месту жительства при наличии полной/частичной ремиссии с рекомендациями;
·частота осмотра пациента один раз в 2-4 недели на первом году, далее – в зависимости от клинического состояния и гематологической динамики, но не реже 1 раза в 3 месяца;
Вакцинация: после окончания иммуносупрессивной терапии при полной/частичной ремиссии вакцинацию проводить в полном объеме, исключив живые вакцины; · снятие пациента с диспансерного учета – реципиенты трансплантации гемопоэтических клеток снимаются с диспансерного учета по окончания иммуносупрессивной терапии, наличии полной ремиссии, отсутствия сопутствующих заболеваний и осложнений трансплантации по истечении 5 лет
14.Лейкозы у детей – эпидемиология, этиология, патогенез, классификация лейкозов у детей и подростков. Клиника, лабораторно-инструментальная диагностика. Маршрутизация детей при подозрении на онкологическую патологию. Основные принципы лечения лейкозов. Осложнения. Исходы. Особенности диспансерного наблюдения.
Лейкоз у детей (лейкемия) – системный гемобластоз, сопровождающийся нарушением костномозгового кроветворения и замещением нормальных клеток крови незрелыми бластными клетками лейкоцитарного ряда.
В детской онкогематологии частота лейкозов составляет 4-5 случаев на 100 тыс. детей. Согласно статистике, острый лейкоз является самым частым онкологическим заболеванием детского возраста (примерно 30%); наиболее часто рак крови поражает детей в возрасте 2-5 лет
На современном этапе доказано этиологическое влияние радиационного излучения, онкогенных вирусных штаммов, химических факторов, наследственной предрасположенности, эндогенных нарушений (гормональных, иммунных) на частоту возникновения лейкоза у детей. Вторичный лейкоз может развиться у ребенка, перенесшего в анамнезе лучевую или химиотерапию по поводу другого онкологического заболевания. (HTLV-I (от англ. human T-cell lymphotropic virus)
На сегодняшней день механизмы развития лейкоза у детей принято рассматривать с точки зрения мутационной теории и клоновой концепции. Мутация ДНК кроветворной клетки сопровождается сбоем дифференцировки на стадии незрелой бластной клетки с последующей пролиферацией.

Клиническая картина ОЛЛ складывается из ряда синдромов.
Интоксикационный - вялость, утомляемость, снижение аппетита, тошнота и рвота, потливость, повышение температуры (за счет выделения опухолевыми клетками пирогенов).
Костно-суставной - оссалгии из-за расширения площади кроветворения (плоские кости), вследствие остеопороза, кортикальной деструкции, периостальных наслоений (трубчатые кости, позвонки).
Анемический –бледность, слабость, утомляемость, систолический шум на верхушке сердца; в анализе крови – нормохромная анемия.
Геморрагический - в результате вторичной тромбоцитопении - петехии, экхимозы на коже и слизистых, кровотечения из слизистых оболочек (тип кровоточивости микроциркуляторный).
Лейкопенический - снижение иммунитета, частые заболевания, присоединение инфекции.
Пролиферативный - увеличение печени, селезенки, лимфоузлов - они обычно плотные, безболезненные (происходит расселение бластных клеток по эмбриональным очагам кроветворения). Лимфоузлы чаще увеличиваются во всех группах, возможно изолированное появление конгломератов, чаще в области шеи.
Особо выделяют нейролейкоз – наличие лейкозной инфильтрации в оболочках головного и спинного мозга, в нервных стволах, ганглиях вегетативной нервной системы. Нейролейкоз является следствием метастазирования бластных клеток в начальной стадии болезни. Метастазирование может происходить двумя путями: контактным (с костей черепа и позвоночника на твердую мозговую оболочку и дуральные воронки черепных и спинальных нервов) и более вероятно диапедезным (из сосудов мягкой оболочки в цереброспинальную жидкость и в вещество мозга). Менингеальная форма проявляется головными болями, тошнотой, рвотой, гиперестезией кожных покровов, положительными менингеальными симптомами. Энцефалитическая - нарушением сознания, судорогами, очаговыми симптомами поражения головного мозга
ОАК – анемия, тромбоцитопения, ускорение СОЭ. Количество лейкоцитов может быть повышенным, сниженным или в норме. В лейкоцитарной формуле - лимфоцитоз, бласты (бластные клетки обнаруживаются не всегда). Между бластными клетками и зрелыми гранулоцитами почти нет промежуточных форм, что отражает провал в кроветворении – лейкемическое зияние. Отмечается гипорегенераторная нормохромная анемия и тромбоцитопения;
Пункция костного мозга с исследованием миелограммы и его клеточности: цитологическое исследование миелограммы – гипер- (нормо- или мало-) клеточный костный мозг, с суженными ростками нормального кроветворения и инфильтрацией бластными клетками – от 25% до тотального замещения костного мозга опухолью.
Для уточнения формы лейкоза проводят морфологическое и цитохимическое исследование клеток костного мозга.
Лечение ОЛЛ направлено на эрадикацию опухолевых клеток, для чего используется полихимиотерапия.
К основным противолейкозным препаратам относятся:
Антиметаболиты (циклоспецифичны, нарушают синтез в основном предшественников нуклеиновых кислот в лейкозных клетках): метотрексат - антагонист фолиевой кислоты, нарушает синтез пуриновых оснований, при введении одинаковой дозы внутриклеточная концентрация метотрексата в 3 раза выше в лимфобластах, чем в миелобластах; 6-меркаптопурин - антагонист пурина, ингибирует синтез пуринов, цитозар - ингибирует синтез ДНК и через 24 часа 90% бластов синхронизируются в S-фазе.
Алкилирующие соединения (нециклоспецифичны) - подавляют синтез ДНК и РНК: циклофосфан (группа азотиприта) - действует цитостатически и цитолитически на клетки в любой фазе митотического цикла.
Алкалоиды (винкристин) - нециклоспецифичен, действует на все фазы, в основном на период митоза.
Ферменты (L-аспарагиназа) - разлагает аспарагин, который не может синтезировать лейкозная клетка, блокирует вступление клеток в период синтеза ДНК.
Антибиотики (рубомицин, даунорубицин) - нециклоспецифичны, подавляют синтез нуклеиновых кислот.
Гормоны (предизолон, дексаметазон) - ингибируют синтез РНК и ДНК в клетке, действуют цитолитически только на лейкозные клетки и не вызывают разрушения нормальных лимфоцитов.
Тактика лечения ОЛЛ:
Максимально переносимые дозы препаратов
Минимальное промежуточное время между введением отдельных доз
Интенсивное лечение побочного действия препаратов
Лечение поражения ЦНС, т.е. профилактика нейролейкемии начинается сразу же после постановки диагноза. Спинномозговые пункции с введением цитостатиков проводятся каждые 2 недели, в конце 3-го протокола назначается дистанционная гамма-терапия в дозе 12-24 Гр (в зависимости от группы риска).
Поддерживающая терапия направлена на уничтожение оставшейся массы лейкозных клеток, проводится в течение 2 лет с использованием 6-меркаптопурина и метотрексата
Вспомогательная терапия включает трансфузионную заместительную терапию, дезинтоксикационное лечение, иммунотерапию, профилактику и лечение инфекционных и других осложнений.
В настоящее время с применением современных программ лечения 5-летняя выживаемость и выздоровление при ОЛЛ у детей достигает 70-90%.
Д-наблюдение детей с острыми лейкозами осуществляется гематологами специализированных центров или кабинетов. Рекомендуется 1 раз в неделю исследовать общий анализ крови с подсчетом тромбоцитов. Контрольные исследования костного мозга по программе BFM в 1-й год ремиссии делаются ежеквартально, на 2-м году 1 раз в 6 месяцев. В детские учреждения больные лейкозом допускаются не ранее, чем через 6 месяцев полной ремиссии (при благоприятном прогнозе этот срок может сокращаться). Ребенка освобождают от занятий физкультурой, проводят профилактику вирусных инфекций. Подход к проведению прививок в настоящее время пересмотрен и решается положительно. Иммунизация живыми вакцинами противопоказана. Пренесшие ОЛЛ имеют право обучаться на дому, освобождаются от службы в армии.
15.Синдром нарушенного кишечного всасывания – целиакия, муковисцидоз, лактазная недостаточность, пищевая аллергия. Эпидемиология, этиология, основные этапы патогенеза заболеваний. Основные клинические синдромы. Лабораторные и инструментальные методы диагностики заболеваний. Роль пренатального и постнатального скринингов в диагностике заболеваний. Значение методов прижизненного морфологического исследования слизистой оболочки кишечника. Принципы лечения заболеваний. Осложнения. Исходы. Профилактика.
Термины «синдром нарушенного кишечного всасывания», или «синдром мальабсорбции», охватывают широкий круг состояний, при которых нарушено усвоение различных питательных веществ.
Синдром недостаточности всасывания - это название любого заболевания, при котором важные питательные вещества (одно или более) или минералы не перевариваются или не поглощаются кишечником должным образом.
Классификация:
1. Первичный синдром мальабсорбции (наследственно обусловленный) возникает при нарушении функции, образовании недостаточного количества или неправильной химической структуры ферментов, принимающих участие в переваривании компонентов пищи, а также нарушении процессов всасывания питательных веществ в кишечнике.
2. Вторичный синдром мальабсорбции (приобретенный): возникает при различных заболеваниях желудка (гастрогенный), поджелудочной железы (панкреатогенный), печени (гепатогенный), тонкого кишечника (энтерогенный), а также послеоперационный, эндокринный, ятрогенный (при длительном применении антибиотиков, слабительных, цитостатиков и других препаратов, лучевой терапии).
Дисахаридазная недостаточность. Заболевание, связанное с дефицитом конкретных дисахаридаз (ферментов) в слизистой оболочке тонкой кишки.
Этиология и патогенез. Известны первичная (наследственная) и вторичная (приобретенная) дисахаридазная недостаточность. Приобретенная недостаточность дисахаридаз может оказаться следствием болезней тонкой кишки (энтерит, язвенный колит, тяжелые кишечные инфекции, целиакия, муковисиидоз).
Клиническая картина. Отмечаются частый жидкий пенистый стул с кислым запахом, срыгивания, рвота, метеоризм, изменение аппетита. Развитие гипотрофии, гипотонии мышц, полигипоавитаминоза и иных дефицитных состояний. Нередко — следствие основного заболевания.
Диагноз. Из лабораторных показателей максимально характерно обнаружение повышенных количеств дисахаридов (лактозы, сахарозы) в кале методом хроматографии или «Clinitest», снижение Рн кала ниже 5,5, резкое уплощение гликемической кривой после нагрузки тем дисахаридом, к которому есть непереносимость (не более 20-25 % от исходного уровня), хотя нагрузочные пробы требуется проводить с огромной осторожностью. Рентгенологическое исследование желудочно-кишечного тракта выявляет избыточное число газа и жидкости в просвете кишки, дискинетические расстройства, чередование участков атонии и спазма, резкое усиление перистальтики.
Прогноз. При правильном лечении благоприятный.
Лечение. Исключение непереносимого дисаха-рида из пищи. При непереносимости лактозы назначается диета с исключением свежего молока и кисломолочных смесей, с введением стандартных низколак-тозных смесей, отмытого от сыворотки творога, сыра. Безмолочный прикорм с ранним введением мясного пюре (с 4-5-го месяца). Обычная продолжительность диеты 6-9-12 месяца, затем дисахаридазная активность у большей части детей восстанавливается. Лечение основного заболевания.
Целиакия – иммуноопосредованное системное заболевание, которое возникает в ответ на употребление глютена или соответствующих проламинов генетически предрасположенными индивидуумами и характеризуется наличием широкой комбинации глютен-зависимых клинических проявлений, специфических антител
Этиология: Глютенэнтеропатия – многофакторная болезнь, но существенная роль в развитии заболевания принадлежит мутации генов. Болезнь носит наследственный характер и передается по аутосомно-доминантному типу.
Виды целиакии
Типичная форма развивается у детей в раннем детском возрасте и характеризуется тремя основными симптомами: нарушением стула, отставанием в росте и увеличением объема живота.
Атипичная, или субклиническая форма – типичные симптомы данной патологии практически отсутствуют, на первое место выходят нарушения со стороны других органов и систем.
Латентная форма – симптомы возникают во взрослом возрасте.
Скрытая форма – протекает бессимптомно.
Рефрактерная форма – обладает устойчивостью к лечению.
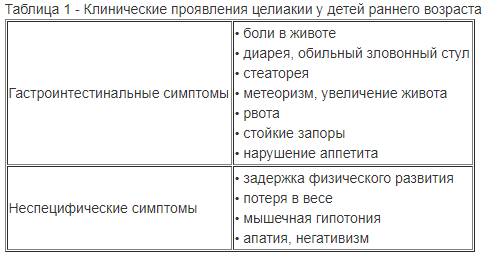
Клиника: Начальные симптомы заболевания могут быть нехарактерными: выраженная слабость, быстрая утомляемость, снижение аппетита. Через два месяца целиакия начинает приобретать характерные симптомы: увеличение размеров живота, вздутие, тошнота, рвота, жидкий стул, чередующийся с запорами, потеря веса.
На 2–3 году жизни при употреблении глютеносодержащих продуктов отмечаются признаки истощения, боль в животе носит тупой, приступообразный характер, проявляются симптомы рахита, задержка прорезывания молочных зубов, отеки, низкорослость.
Порой глютеновая энтеропатия проявляется нетипично. Тогда могут возникать воспалительные поражения полости рта, артриты крупных суставов, анемии, дерматиты, жажда и обильное выделение мочи.

Жалобы и анамнез
В типичном случае целиакия манифестирует через 1,5 – 2 месяца после введения в рацион питания ребенка глютенсодержащих продуктов (сухарики, хлеб, сушки, баранки, манная (пшеничная) каша, мультизлаковая каша). Иногда манифестация целиакии у детей происходит после перенесенных инфекционных заболеваний (кишечных или респираторных инфекций), однако часто заболевание начинается без видимой причины.
Клинические симптомы целиакии появляются, в большинстве случаев, постепенно. Появляется свойственный для целиакии обильный пенистый, жирный, зловонный стул, нарушение аппетита, беспричинная рвота, потеря массы тела. Родители обращают внимание на нарушения поведения – появляется раздражительность, негативизм, апатия, нарушается сон, исчезает интерес к окружающему.
Старшие дети жалуются на боли в животе, которые чаще имеют непостоянный, «тупой» характер и локализуются преимущественно в околопупочной области.
Диагностика целиакии
Наиболее специфичной методикой выявления целиакии является определение в крови антител к глиадину и антител к тканевой трансглутаминазе. Чувствительность методики составляет 100%, ее специфичность для данной патологии порядка 95-97%.
Помимо этого, можно произвести биопсию слизистой тонкого кишечника и определить имеющуюся атрофию (сглаживание) ворсин, а также наличие скоплений лимфоцитов в слизистой.
Дополнительными методиками для уточнения состояния больного являются эндоскопическое исследование тонкого кишечника, тест Шиллинга и проба с D-ксилозой, УЗИ органов брюшной полости, компьютерная томография, МРТ-ангиография мезентериальных сосудов, контрастная рентгеноскопия кишечника.
Лечение:
Единственным методом лечения целиакии является безглютеновая диета. Детям младшего возраста назначают специальные смеси, в составе которых нет глютена. Из рациона исключаются продукты, содержащие пшеницу, рожь, ячмень и овес. Разрешено употреблять мясо, рыбу, птицу, овощи, фрукты, яйца и молочные продукты, рис, гречку.
При соблюдении диеты симптомы заболевания исчезают через полгода.
Медикаментозная терапия заключается в использовании витаминов и пищеварительных ферментов. При тяжелом течении болезни проводится заместительная терапия: внутривенно вводятся аминокислоты, жировые смеси и глюкоза.
Исходы
Если больной придерживается диеты, то прогноз благоприятный. Несвоевременная диагностика или погрешности в рационе ведут к прогрессированию заболевания и увеличивают риск возникновения осложнений:
развитие язв тонкой кишки;
гиповитаминоз;
задержки в психическом и умственном развитии;
остеопороз и переломы костей;
при стремительном течении заболевания у детей младше двух лет возможен летальный исход
Срок наблюдения: пожизненно. Ведение больного осуществляет детский гастроэнтеролог и сертифицированный диетолог.
Кратность наблюдения: после установки диагноза в течение первых двух лет – 1 раз в 6 месяцев, с 3-го года наблюдения при условии установления стойкой ремиссии и регулярных достаточных весоростовых прибавок – 1 раз в год.
проведение профилактических прививок в период ремиссии.
Муковисцидоз см выше(кишечная форма была уже)
Пищевая аллергия (ПА) - это вызванная приемом пищевого продукта патологическая реакция, в основе которой лежат иммунные механизмы (специфические IgE-опосредованные реакции, клеточный иммунный ответ (не IgE-опосредованные) или их сочетание - реакции смешанного типа)
Причины - прием белков и т.п. (пищевых аллергенов)
Пищевые аллергены - любые вещества, чаще всего белковой природы, стимулирующие выработку IgE или клеточный иммунный ответ. В так называемую «большую восьмерку» продуктов, наиболее часто вызывающих аллергические реакции, входят: коровье молоко, куриное яйцо, арахис, орехи, рыба, морепродукты, пшеница и соя.
Современная классификация проявлений ПА основана на клинико-иммунологическом принципе. Выделяют следующие клинические проявления ПА:
IgE-опосредованные реакции
• Оральный аллергический синдром (пищевая аллергия, обусловленная сенсибилизацией к пыльце)
• Крапивница/ангиоотек
• Риноконъюнктивит/астма
• Гастроинтестинальные симптомы (тошнота, рвота, боли в животе и диарея)
• Анафилаксия
• Анафилаксия при пищевой аллергии, индуцированная физической нагрузкой
Смешанные IgE-опосредованные и клеточные реакции
• Атопический дерматит 12
• Эозинофильная гастроинтестинальная патология
Проявления, опосредованные клеточными реакциями
• Индуцированный пищей проктит, проктоколит, энтероколит
• Индуцированная пищей энтеропатия
Клинические проявления ПА и возраст манифестации варьируют в зависимости от характера реакции.
Оральный аллергический синдром (пищевая аллергия, обусловленная сенсибилизацией к пыльце) – характерен зуд, легкий отек ограничивается полостью рта. Начало проявлений после установления поллиноза. Возможно как персистирование, так и зависимость от сезона.
Крапивница/ангиоотек, возникающие при приеме внутрь или при контакте с пищевым продуктом.
Риноконъюнктивит/астма - редкие проявления ПА, возможны при вдыхании аэрозоля аллергена. У младенцев и детей встречается чаще, чем у взрослых
Гастроинтестинальные симптомы - тошнота, рвота, боли в животе и диарея, вызванные приемом пищи
Анафилаксия - быстрая прогрессирующая мультисистемная реакция 13 Анафилаксия при пищевой аллергии, индуцированная физической нагрузкой - пища провоцирует анафилаксию только в случае дальнейшей физической нагрузки
Атопический дерматит - ассоциируется с пищевой аллергией у 30-40% детей со среднетяжелым и тяжелым АтД, у детей младшего возраста- чаще.
Эозинофильная гастроинтестинальная патология - симптоматика зависит от уровня ЖКТ, вовлеченного в процесс и степени эозинофильного воспаления.
Индуцированный пищей проктит, проктоколит, энтероколит - характерна слизь и кровь в стуле. Преимущественно встречается у младенцев и обычно разрешается к более старшему возрасту
Индуцированная пищей энтеропатия. Хронические проявления: рвота, диарея, отставание в росте, вялость. При повторном введении продукта после элиминации характерны: рвота, диарея, гипотензия в течение 2 ч после приема. Преимущественно встречается у младенцев и обычно разрешается к более старшему возрасту.
Диагностика: Определение уровня специфических антител класса IgE
Кожное тестирование позволяет подтвердить наличие сенсибилизации и эффективно в диагностике IgE-опосредованной ПА
диагностическая элиминационная диета
Этиологическим лечением является исключение из питания причинно-значимых продуктов.
+ антигистаминные по показания
Больные с легкими проявлениями ПА могут наблюдаться амбулаторно, консультации специалистов (в зависимости от характера проявления и по показаниям – аллерголога, диетолога, гастроэнтеролога, дерматолога) с частотой 1 раз в 2-6 месяцев. При тяжелых и среднетяжелых реакциях на пищу ребенок может нуждаться в госпитализации для обследования, подбора терапии и коррекции рациона, реабилитационных мероприятий (1 раз в 3-12 мес., в зависимости от характера патологических проявлений).
16.Врожденные и наследственные заболевания почек (синдром Альпорта, врождѐнный нефротический синдром, болезнь тонких базальных мембран). Наследственные болезни, их отличие от врожденных. Понятие о методах исследования наследственной патологии. Этиология и основные этапы патогенеза врожденных и наследственных заболеваний почек. Основные клинические синдромы. Лабораторные и инструментальные методы диагностики заболеваний. Дифференциальный диагноз. Исходы. Лечение. Прогноз.
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.

патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).







Болезнь тонких базальных мембран (БТБМ) (синонимы: семейная доброкачественная гематурия) - генетически детерминированная неиммунная гломерулопатия, связанная с мутацией генов коллагена IV типа COL4A4/COL4A3, проявляющаяся гематурией, в большинстве случаев не имеющая тенденции к прогрессированию, поэтому нередко называемая «семейной доброкачественной гематурией».
Клиника. Основным клиническим проявлением БТБМ является гематурия. Гематурия наблюдается пожизненно, при этом, как правило, не отмечается снижения почечных функций.
Диагностика БТБМ базируется на двух критериях: 1) гематурия в семье; 2) специфические изменения БМ клубочков в биоптате почки. Основа диагностики-биопсия
При решении вопроса о терапевтической тактике у пациента с БТБМ целесообразно исследование функционального состояния почек. При нормальных показателях функции необходимо динамическое наблюдение за состоянием ребенка с повторными функциональными пробами.
Основные принципы построения лечебных программ:
сбалансированное по основным градиентам питание;
лечебная физкультура;
ограничение контактов с инфекционными больными;
санация очагов инфекции;
вакцинация по индивидуальному плану;
мембраностабилизирующая, антиоксидантная терапия (димефосфон, эссенциале, витамины группы А, Е, В6, и т.д.);
энерготропная терапия (элькар, кудесан);
сеансы гиперболической оксигенации;
ренопротективная, антипротеинурическая, антисклеротическая терапия ингибиторами АПФ (каптопри, энап, моноприл и др.).
Нефротический синдром — клинико-лабораторный симптомокомплекс, характеризующийся: протеинурией, гипопротеинемией, гипоальбуминемией, диспротеинемией; гиперлипидемией, липидурией; отеками периферическими, полостными и степени анасарки.
Врожденный нефротический синдром – НС, развившийся у детей с момента рождения или в первые 3 месяца жизни. У детей он гетерогенен, выделяют первичные и вторичные формы врожденного НС.
А. Первичные формы:
- Врожденный НС финского типа
- Врожденный НС французкого типа
- Другие НС (с минимальными изменениями, ФСГС, мембранозный ГН)
- Синдромальные аномалии (синдром Галловей-Моуат, врожденный НС с аномалиями нервной системы и другие синдромы)
В. Вторичные формы:
- На фоне инфекционных заболеваний (врожденный сифилис, токсоплазмоз, краснуха, цитомегалия, малярия)
- При СКВ у матери
- НС ассоциированный с тромбозом почечных вен
чаще финского типа. - нефротический синдром с гематурией
Жалобы на появление отеков и уменьшение количества мочи + ОАК, ОАМ,
Прогноз без трансплантации почек крайне не благоприятный
Цель – довести ребенка до возраста, приемлемого для трансплантации, которая является единственным средством излечения. Протокол, предложенный финскими педиатрами в начале 90-х г. включает:
1) Компенсацию гипоальбуминемии (20% альбумин) в сочетании с фуросемидом до уровня альбумина сыворотки 15-20 г/л.
2) Заместительную терапию (витамин Д, тироксин, витамины, кальций).
3) Питание (энтеральное через назогастральный зонд 130 ккал/кг, 4 г/кг/сут белка, жидкость 100-130 мл/кг/сут; 10-14% белок, 40-50% липиды, 40-50% углеводы).
4) Профилактику и лечение тромботических осложнений (курантил, гепарин, низкомолекулярные гепарины).
5) Применение ингибиторов АПФ (капотен).
6) Профилактику и лечение инфекционных осложнений
Наследственные болезни — заболевания, возникновение и развитие которых связано с изменениями (мутациями) генетического материала.
Врожденная патология в виде врожденных пороков развития может возникнуть в критические периоды внутриутробного развития под действием факторов внешней среды (физических, химических, биологических и др.). При этом поражения или изменения генома нет.
17. Дыхательная недостаточность у детей – классификация, виды нарушения функции внешнего дыхания, причины их развития. Патогенез развития вентиляционной обструктивной дыхательной недостаточности. Клинические проявления вентиляционной обструктивной дыхательной недостаточности у детей. Методы исследования функции внешнего дыхания. Принципы лечения острой дыхательной недостаточности вентиляционной обструктивной. Показания для перевода на ИВЛ. Патогенез формирования легочного сердца и лѐгочно-сердечной недостаточности при заболеваниях лѐгких у детей. Принципы лечения хронической дыхательной недостаточности.
Острая дыхательная (респираторная) недостаточность (ОДН) – быстро нарастающее (время развития несколько минут/дней) тяжелое патологическое состояние больного, обусловленное несоответствием системы внешнего дыхания метаболическим потребностям организма для поддержания нормального парциального напряжения кислорода и углекислого газа в артериальной крови, или оно достигается за счет усиленной работы систем дыхания и кровообращения, что приводит к снижению и последующему истощению функциональных возможностей организма.
Патогенетическая классификация ОДН (Шанин Ю.Н., Костюченко А.Л., 1975): диффузионная, вентиляционная, смешанная.
Гипоксемическая дыхательная недостаточность (диффузионная, паренхиматозная, легочная, ДН 1-го типа) характеризуется артериальной гипоксемией, развивающейся главным образом в результате нарушения регионарного вентиляционно-перфузионного баланса или внутрилегочного шунтирования крови.
Нарушение газообмена в легких
Основными патогенетическими механизмами гипоксемии являются:
• Снижение парциального давления кислорода во вдыхаемом воздухе;
• Общая гиповентиляция легких;
• Нарушения диффузии газов через альвеолокапиллярную мембрану;
• Нарушение вентиляционно-перфузионных отношений;
• Шунт (прямой сброс венозной крови в артериальную систему кровообращения);
• Снижение парциального напряжения кислорода в смешанной венозной крови.
Вентиляционная дыхательная недостаточность (гиперкапническая, «насосная», ДН 2-го типа) обусловлена первичным уменьшением эффективности легочной вентиляции (альвеолярная гиповентиляция), что нарушает выведение СО2 и нередко приводит к нарушениям кислотно-основного состояния (КОС), т.е. характерным признаком является гиперкапния (PaCO2≥ 45 мм рт.ст.), гипоксемия также присутствует, но хорошо поддается терапии кислородом. Уровень гиперкапнии прямо пропорционален степени уменьшения альвеолярной вентиляции.
Патофизиологические механизмы гиперкапнии:
• снижение минутной вентиляции легких (гиповентиляция);
• увеличение физиологического «мертвого» пространства;
• повышение продукции углекислоты.
При данном типе ОДН снижена альвеолярная вентиляция. За единицу времени в альвеолы поступает меньше воздуха, чем в норме: а) рестриктивные нарушения – дыхательная поверхность и эластичность легких уменьшены; б) обструктивные нарушения – нарушения бронхиальной проходимости, спазм бронхов, гиперсекреция слизи, отек слизистой оболочки бронхов; в) расстройства нервной регуляции - гиповентиляция при поражении дыхательного центра или периферических нервов.
Смешанная дыхательная недостаточность. Нарушено как распределение газа в легких (вентиляционно-перфузионные отношения, так и вентиляционная (насосная) функция легких.
Клиника:
-нарушения сознания (спутанность, заторможенность вплоть до комы или возбуждение); см. шкалу ГЛАЗГО;
- одышка или удушье (увеличение частоты дыхания: ЧД более 24 в1 мин. является признаком ОДН; ЧД 27±5 в 1 мин. указывает на тяжелую ДН; при крайне тяжелой ОДН ЧД превышает 35 в 1 минуту; ЧД 12 и менее является предвестником остановки дыхания.
-цианоз кожи и слизистых оболочек;
- повышенная потливость;
- тахикардия или сердечные аритмии;
- артериальная гипертензия;
- артериальная гипотензия (в очень тяжелых случаях).
По уровню декомпенсации систем дыхания и кровообращения, сознания, содержанию кислорода и углекислоты в крови различают III стадии ОДН.
I стадия ОДН. Пациент находится в сознании, жалуется на чувство нехватки воздуха, беспокоен, астеничен. Кожные покровы бледные, влажные, небольшой акроцианоз видимых слизистых. ЧД до 30 в 1 мин, ЧСС до 110 в 1 мин, АД в норме или несколько повышено, paO2 снижается до 60-70 мм рт.ст (90-94%)., paСO2 снижено из-за компенсаторной одышки. (Симптомы при нагрузке) - Будесонид/пульмикорт 1мг/инг
II стадия ОДН. Больной жалуется на выраженное удушье, возможно развитие психомоторного возбуждения, нарушение сознания, бреда, галлюцинаций. Кожные покровы влажные, цианотичные, нередко в сочетании с гиперемией. ЧД 30-40 в 1 мин, ЧСС 120-140 в 1 мин нередко аритмия, регистрируется гипертензия, paO2 снижается 45-59 мм рт.ст. (75-89%), paСO2 повышается до 50 мм рт.ст. (Симптомы в покое) - Сразу 2 мг/инг, иначе дексаметазон 0.6мг/кг + госпитализация
III стадия ОДН. Сознание помрачено или отсутствует, возможно развитие судорожного синдрома из-за гипоксии мозга, наблюдается пятнистый цианоз, гипоксическое расширение зрачка с отсутствием реакции на свет. При прогрессировании процесса тахипное (ЧД > 40 в 1 мин) переходит в брадипное (ЧД < 8 в 1 мин). Наблюдается гипотензия, тахиаритмии, paO2 уменьшается менее 40 мм рт.ст. (менее 75%) и ниже, paСO2 повышается до 90 мм рт.ст. и выше.
Основные принципы лечения ОДН:
оказание медицинской помощи, направленной на восстановление проходимости дыхательных путей, нормализацию газообмена и легочной вентиляции;
определение и устранение главных причин развития синдрома ОДН;
устранение нарушений системы кровообращения;
симптоматическая терапия, направленная на коррекцию КОС, обезболивание, устранение гипо- или гиперволемии и пр.
Интенсивное лечение синдрома ОДН начинают с кислородотерапии (увлажненный 40% теплый)
Жаропонижающие препараты при лихорадке выше 39 °С
Ненаркотические анальгетики для обезболивания при плевральной боли (кеторолак в/в 30 мг/1 мл)
При бронхообструктивном синдроме – бронходилататоры (сальбутамол ингаляционно 1-2 дозы/100—200 мкг аэрозоля или 2,5-5 мг через небулайзер).
Дезинтоксикационная терапия (изотонический раствор, 5% раствор глюкозы, гемодез-Н, объём однократного введения 200 – 400 мл).