

К оньяк расширяет сосуды, а виски - связи.
1.Общая характеристика демиелинизирующих заболеваний
Демиелинизирующее заболевание представляет собой патологический процесс разрушения миелиновой оболочки, при котором поражаются нейроны головного и спинного мозга. При этом ухудшается проводимость импульсов в нервной системе. Болезнь характеризуется уничтожением миелина мозга. Это опасное состояние влияет на функционирование всего организма. Заболевание встречается с одинаковой частотой как у взрослых, так и детей. Современная медицина не обладает средствами для полного излечения от этого заболевания. Его можно лишь ослабить и замедлить течение.
Классификация демиелинизирующих заболеваний нервной системы
I Демиелинизирующие заболевания с преимущественным поражением центральной нервной системы.
1. Острые формы.
Первичные (острый рассеянный энцефаломиелит; клинические формы – энцефаломиелополирадикулоневрит, оптикоэнцефаломиелит, оптикомиелит, полиоэнцефалит, диссеминированный миелит);
Вторичные (параинфекционные – энцефаломиелит при кори, коклюше, ветряной оспе, гриппе, герпесе и др.; вакцинальные – при вакцинации АКДС, КДС, антирабической вакциной).
2. Подострые формы. Рассеянный склероз, клинические формы – цереброспинальная, спинальная, церебральная, оптическая, стволовая, мозжечковая.
3. Хронические формы. Энцефалит Ван Богарта, Даусона, Петте, Деринга, периаксиальный диффузный лейкоэнцефалит Шильдера.
II Демиелинизирующие заболевания с преимущественным поражением периферических нервов.
Инфекционно-аллергический первичный полирадикулоневрит Гийена-Барре, инфекционные полирадикулоневропатии (дифтерийная), токсические полинейропатии, дисметаболическая и диабетическая полинейропатия.
2. Общая характеристика рассеянного склероза
Рассеянный склероз - заболевание нервной системы, возникающее в молодом и среднем возрасте (15–40 лет). Особенностью болезни является одновременное поражение нескольких различных отделов нервной системы, что приводит к появлению у больных разнообразной неврологической симптоматики. Еще одна особенность заболевания – ремиттирующее течение.
3. Эпидемиология рассеянного склероза

4. Этиология рассеянного склероза
Этиология. РС считается полиэтнологячным заболеванием. Существует несколько теорий об этиологии РС.
-Вирусная инфекция (ретровирусы, человеческий вирус герпес-б, парамиксавирусы - корь, собачья чумка; коронавирусы, аденовирусы и др.) повреждает олигодендроглию, далее на продукты распада миелина и на сами вирусы запускается иммунная реакция , что и вызывает обострение клинических проявлений.
-Бактериальная инфекция имеет сходные антигены; под влиянием высокой температуры белки приобретают необратимые изменения и запускают каскад цитокиновых реакций, которые приводят к демиелинизации.
5. Патогенез рассеянного склероза

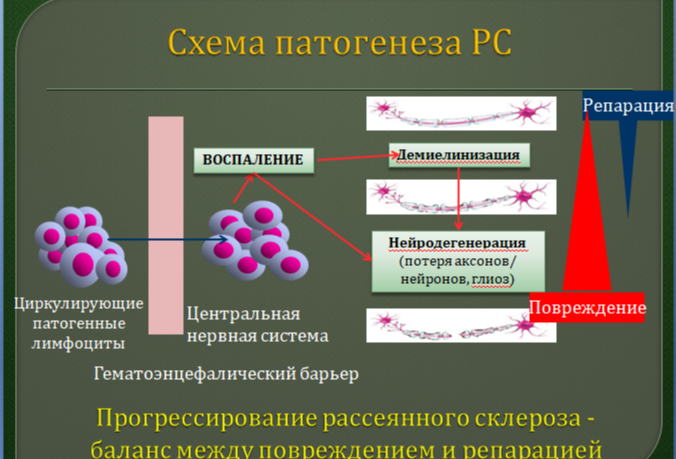
6. Морфология рассеянного склероза

: в раннюю стадию формирования склеротической бляшки в нервной ткани вокруг сосудов обнаруживаются лимфоциты и мононуклеарные клетки, начинается демиелинизация периваскулярной области. иелиновые оболочки набухают, образуя утолщения по ходу волокон, затем начинают распадаться. Аксоны имеют неравномерную толщину, с утолщениями и истончениями. Продукты распада миелина утилизируются клетками микроглии, наблюдается их увеличение и зернистость. При старении бляшки ее инактивация начинается из центра; по краям еще наблюдаются лейкоциты, в центре происходит замещение нервной ткани соединительной. Старая неактивная бляшка лишена олигодендроглии и состоит из соединительной ткани [1]. еории возникновения рассеянного склероза (РС) можно разделить на две группы по предполагаемым провоцирующим факторам. Эндогенные: -теория (некоторые вариации в A-комплексе, в частности, в генах Q1 и R1, в 6 хромосоме увеличивают риск возникновения РС); теория сбоя в работе тромбоцитов (повышенная активность тромбоцитов у пациентов с РС вызывает выброс моноцитами I1-a, который активирует клетки эндотелия и может выступать медиатором в тромбоцит-связанной трансэндотелиальной инфильтрации лейкоцитов в периферические ткани). Экзогенные: теория недостатка витамина (недостаток витамина в раннем возрасте может приводить к нарушениям в активации и пролиферации лимфоцитов и дифференцировке -хелперов, а также развитии нервной ткани); теория кишечной микробиоты (дисбиоз кишечника оказывает влияние на развитие и созревание иммунных клеток и приводит к дисбалансу про- и противовоспалительных цитокинов в кишечнике и организме в целом, и также опосредованно может влиять на целостность ГЭБ) [2]; теория хронических вирусных инфекций (некоторые распространенные ДНК-вирусы способны проникать в ЦНС и сохраняться в ней пожизненно; сходства протеинов эпитопов этих вирусов с и P могут служить основой аутоиммунной реакции) [3]. Выводы: морфологически рассеянный склероз проявляется в демиелинизации нервных волокон, сопровождающейся местным воспалением и последующей заменой нормальной нервной ткани на соединительную. РС — аутоиммунное заболевание, вероятно, провоцируемое некими факторами внешней или внутренней среды или их сочетанием. Решающий фактор в настоящее время не выяснен.
7. Критерии диагностики рассеянного склероза


8. Классификация форм рассеянного склероза
Классификация рассеянного склероза.
1. По уровню поражения:
- церебральная
- спинальная
- цереброспинальная
-оптическая
-стволовая
2. По течению:
- ремитирующая
- первично-прогрессирующая
- вторично-прогрессирующая
9. Двигательные Чувствительные Координаторные Зрительные
Клиническая картина. Заболевают преимущественно в возрасте от 15 до 40 лет, реже - в детском и пожилом возрасте. Наиболее часто первыми симптомами заболевания являются признаки поражения зрительного нерва (ретробульбарный неврит): ощушение нечеткости изображения, преходящая слепота, снижение остроты зрения, скотомы. Заболевание может начинаться глазодвигательными расстройствами (диплопия, косоглазие), нестойкими пирамидными симптомами (центральный моно-, геми- или параларез с высокими глубокими рефлексами, клонусами стоп, патологическими стопными и кистевыми знаками), мозжечковыми нарушениями (шаткость при ходьбе, интенционное дрожание), расстройствами чувствительности в конечностях (онемение, парестезии).
Значительно реже первыми признаками болезни могуr быть нарушения функции тазовых органов (задержка мочеиспускания, императивные позывы), вегетативно-еосудистая дистония, поражения лицевого, тройничного нервов и нервов бульбарной группы. У женщин может нарушаться менструальный цикл, у мужчин развивается импотенция.
Характерным ранним (но не обязательным) признаком заболевания является снижение или исчезновение брюшных рефлексов. В отличие от других заболеваний нервной системы при повторных обострениях болезни возникают новые симптомы. Нарушения когнитивных функций появляются в более поздних стадиях болезни, чаще в виде эмоциональной неустойчивости, эйфории или депрессии, раздражительности, вялости, апатии, снижения интеллекта различной степени вплоть до деменции. Эпилептические припадки при рассеянном склерозе наблюдаются редко.
Цереброспинальная форма, характер изующаяся многоочаговостью поражения уже в начальной стадии болезни, симптомами поражения координаторных и пирамидных систем в головном и спинном мозге, а также зрительных, глазодвигательных, вестибулярных и других систем.
Мозжечковая форма чаще проявляется симптомами поражения ствола мозга и мозжечка, реже - только мозжечковыми симптомами: скандированная речь, горизонтальный, вертикальный, ротаторный крупноразмашистый нистагм, адиадохокинез, дисметрия , атаксия, интенционное дрожание в верхних и нижних конечностях, расстройства почерка
Стволовая форма РС с быстро проrрессирующим течением и даже летальным исходом. На фоне головной боли с рвотой выявляются симптомы нарушения функции ствола мозга и мозжечка.
При оптической форме ведущим клиническим симптомом является снижение остроты зрения, которое восстанавливается через некоторое время самостоятельно или на фоне леченияПри офтальмоскопии выявляются признаки ретробульбарного неврита: побледнение диска зрительного нерва.
Спинальная форма характеризуется симптомами поражения спинного мозга на различных уровнях. Ведущими в клинической картине являются нижний спастический парапарез, проводникавые нарушения чувствительности, различной степени выраженности тазовые расстройства.
13.Варианты течения рассеянного склероза
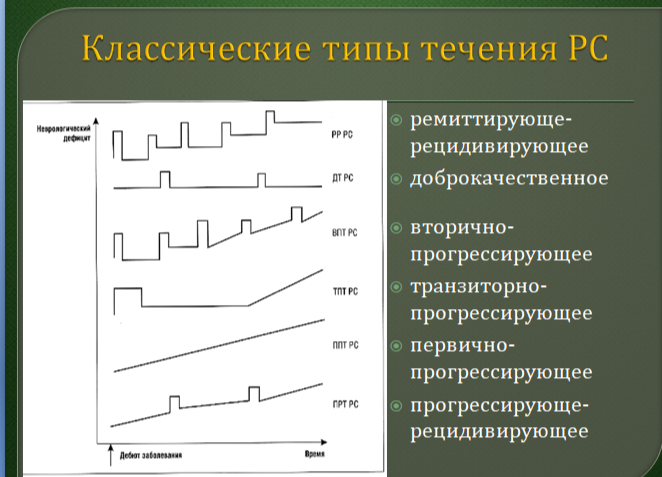
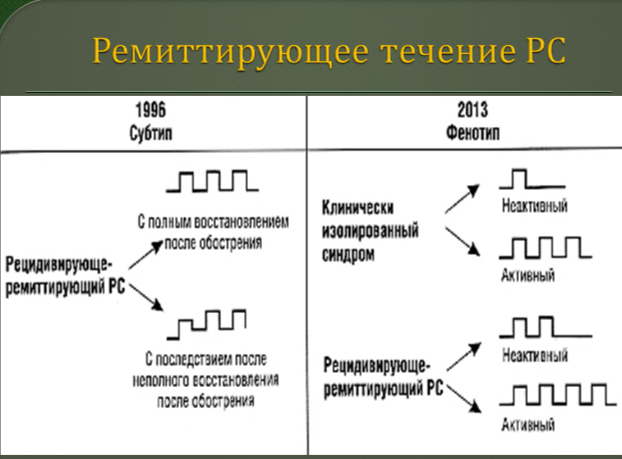
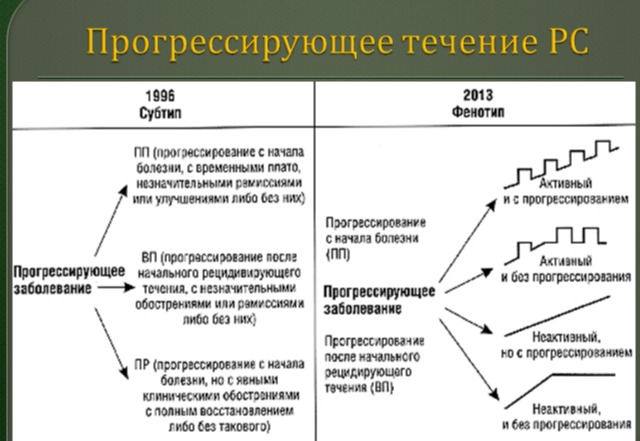
14. Фазы развития рассеянного склероза
Начальная. Обычно проявляется первыми обострениями, которые могут быть непродолжительными и менее интенсивными. Первым клиническим признаком возможного рассеянного склероза становится мышечная слабость. В этот период симптоматика незначительная, проходит бесследно и не беспокоит пациента. 1
Активная. На этом этапе появляются более частые и интенсивные обострения. Симптомы могут становиться более выраженными и разнообразными. 1
Прогрессирующая. Характеризуется постоянным ухудшением состояния, независимо от обострений. Может наблюдаться нарастание инвалидизации. 1
Поздняя. На данной стадии симптомы становятся постоянными и выраженными, способствуя значительным ограничениям в повседневной жизни. Часто требует постоянной медицинской и социальной поддержки.
15. Осложнения РС
Прогрессирование патологии способно приводить к инвалидизации людей с этим диагнозом. В зависимости от того, насколько своевременно было начато лечение, ухудшение ситуации может иметь сроки от 5 до 30 лет. В среднем длительность жизни с признаками рассеянного склероза сокращается на 15 лет. К осложнениям этого заболевания относят такие нарушения:
потерю чувствительности конечностей;
частичный или полный паралич;
значительную утрату зрения и слепоту;
потерю контроля над мочеиспусканием и дефекацией;
судороги;
умственные расстройства.
Нарушения работы ЦНС негативно сказываются на состоянии всего организма. Замедление передачи сигналов ухудшает контроль больного над работой внутренних органов, поэтому можно вовремя не заметить те или иные расстройства. То же касается и функционирования мозга. Ухудшение реакций повышает риск падений, ожогов, обморожений, получения иных травм.
16. Клинико-лабораторные и биохимические изменения при РС
Диагностика. Диагноз РС устанавливают на основании характерных клинических синдромов и их динамики: нистагм, скандированная речь и интенционный тремор (триада Шарко), а также отсутствие брюшных рефлексов, побледнение височных половин дисков зрительных нервов и задержка мочеиспускания (пентада Марбурга). Диагностически важным является симптом Лермитта - ощущение прохождения электрического тока по спине и конечностям при наклоне головы кпереди, а также иреходящее нарастание выраженности двигательных расстройств при повышении температуры окружающей среды (симптом горячей ванны)
Лабораторные исследования:
• ОАК: норма или небольшие воспалительные изменения;
• ОАМ: при наличии расстройств мочеиспускания: увеличение количества лейкоцитов, появление белка в моче;
• Анализ ликвора: лимфоцитарный плеоцитоз и увеличение иммуноглобулинов G в ликворе;
• лимфоцитарный плеоцитоз и увеличение иммуноглобулинов G в ликворе.
17. Иммунологические изменения при РС
иммунологические показатели, среди которых выделяются: 1 – повышение уровня γ–глобулинов в спинно–мозговой жидкости (СМЖ) (у 75% больных); 2 – выявление олигоклональных иммуноглобулинов класса G (IgG) в СМЖ (у 85–90% больных) уже на ранней стадии заболевания; 3 – повышенное содержание в СМЖ основного белка миелина, что может служить признаком обострения процесса; значения этого показателя более 9 нг/мл свидетельствуют об активной демиелинизации. В диагностических критериях РС [МакДональд и др., 2005 г.] присутствует пункт на исследование ликвора (олигоклональные группы IgG или повышенный индекс IgG). Обнаружение олигоклонального IgG в ликворе с помощью изоэлектрофокусирования, ассоциированного с иммунодетекцией, признано наиболее чувствительным и информативным иммунологическим методом диагностики РС, является своего рода «золотым стандартом» иммунологической диагностики РС [1]. Следует подчеркнуть, что выявление олигоклональных групп IgG в ликворе не является строго специфичным для РС тестом. Эти IgG могут быть выявлены у больных с различными воспалительными заболеваниями мозга, неврологическими проявлениями СПИДа и другими заболеваниями. В то же время около 5% больных с клинически достоверным РС могут не иметь олигоклональные группы IgG в ликворе, а специфичность олигоклональных Ig может меняться по мере развития заболевания. При получении результатов анализа ликвора необходимо также обращать внимание на следующее [1]: – у подавляющего числа больных РС общий белок ликвора составляет менее 70 мг/дл (более высокая концентрация белка обычно свидетельствует в пользу другого заболевания); – цитоз обычно не превышает 30/мм3; – 90% клеток ликвора относятся к лимфоцитам и Т–клетки составляют почти 90% из них. Для оценки характера течения РС используются менее инвазивные и более доступные методы (материалом для исследования является периферическая кровь) [1]: 1) цитофлюориметрический или иммунофлюоресцентный методы определения субпопуляций Т– и В–лимфоцитов с помощью наборов моноклональных антител (МАТ) CD3+, CD4+, CD8+, CD20+, CD25+ и др.; 2) метод оценки пролиферации лимфоцитов: спонтанной и под действием Т– и В–клеточных митогенов (ФГА, Кон–А, PWМ) и ОБМ – реакция бласттрансформации лимфоцитов; 3) метод определения активности супрессорных лимфоцитов: спонтанной и Кон–А индуцированной; 4) метод определения чувствительности иммунокомпетентных клеток к нейроспецифическим антигенам – белку S–100, антигену нейрональных мембран, основному белку миелина, галактоцереброзам и др. – реакция торможения адгезии лейкоцитов; 5) метод определения кислородзависимой бактерицидности фагоцитирующих клеток периферической крови в тесте восстановления нитросинего тетразолия (НСТ–тест); 6) метод радиальной иммунодиффузии в геле для определения количества иммуноглобулинов классов A, G, M. В последние годы с помощью иммуногистохимических и иммуноцитохимических методов получены новые патоморфологические данные, указывающие на наличие патоморфологических вариантов PC. На практике эта неоднородность заболевания может проявляться еще одним сложным аспектом: разным ответом (или отсутствием ответа) на проводимое лечение. В этих условиях целесообразен поиск показателей, характеризующих различные «варианты» PC и позволяющих прогнозировать эффект лечения. Улучшение понимания патогенеза РС и стремление изменить течение заболевания привело к созданию парентеральных и таблетированных иммуномодулирующих и иммуносупрессивных препаратов, изменяющих течение РС (ПИТРС): интерферонов бета, глатирамера ацетата, митоксантрона, Гилении, мовектро, натализумаба, которые достоверно снижают частоту обострений и замедляют нарастание инвалидизации, влияют на активность иммунопатологического процесса по данным МРТ. Однако иммуносупрессорный механизм действия и связанные с этим побочные эффекты ряда новых ПИТРС предполагают существенное повышение значимости гематологических иммунологических методов обследования как при назначении иммуносупрессоров, так и при длительной терапии РС. Лечение РС является одной из наиболее актуальных и сложных проблем современной неврологии. Имеющиеся в распоряжении врачей препараты могут лишь снизить частоту и тяжесть обострений, а также замедлить темпы накопления неврологического дефицита, что подтверждается позитивной динамикой по данным МРТ головного и спинного мозга [8,9,12,13,18,22,23]. Одной из ключевых проблем лечения РС является создание и клиническое исследование новых препаратов, блокирующих развитие атрофии головного мозга, воспаления и демиелинизации, снижающих частоту обострений, предотвращающих прогрессирование инвалидизации. Обнадеживающие результаты последних исследований эффективности патогенетической терапии фокусируют внимание властей, общественности, больных и специалистов на новые возможности лечения и обеспечения больных РС, дают новые надежды больным и членам их семей, особенно в случае ремиттирующего варианта течения заболевания. Современным стандартом лечения обострения заболевания средней и значительной степени тяжести является высокодозная пульс–терапия метилпреднизолоном по 1 г ежедневно в течение 3–5 дней, внутривенно, капельно. Возможно внутримышечное введение дексаметазона в течение 7–14 дней или прием преднизолона перорально сроком до 30 дней [1,21]. Для профилактики обострений и предотвращения прогрессирования заболевания в настоящее время повсеместно используются 5 препаратов из группы ПИТРС (препараты, изменяющие течение РС)
18. Дополнительные методы диагностики РС
Магнитно-резонансная томография головного и спинного мозга. Этот метод считается наиболее прогрессивным и информативным, не причиняет дискомфорта пациенту и не имеет побочных действий. Цена обследования — от 4 000 рублей, точность превышает 95-96 %.
Спинномозговая пункция. Процедура заключается во взятии образца из позвоночного канала. Забор выполняется под общим наркозом. Метод позволяет определить состояние нервных волокон. Цена — от 16 000 рублей, точность — до 98 %.
Измерение вызванного потенциала. Аппаратный метод, который заключается в отслеживании электрических импульсов, вырабатываемых мозгом. Он отличается безболезненностью и быстрым получением результатов. Цена — от 6 000 рублей, точность результатов — от 85 %.
19. Дифференциальная диагностика РС
Таблица 2. Заболевания, сходные с рассеянным склерозом по клиническим и МРТ-критериям:
Заболевание |
Исследования для подтверждения диагноза |
Острый рассеянный энцефаломиелит (ОРЭМ) |
МРТ: один большой очаг (1-2 см в диаметре), очаги множественные, часто сливные и с положительным масс-эффектом, могут быть в базальных ганглиях, таламусе, сером веществе. Располагаются супра и/или инфратенториально. Необязательное накопление контраста во всех очагах (одновременное накопление контраста в нескольких очагах увеличивает вероятность ОРЭМ) |
СПИД |
Антитела к ВИЧ в сыворотке |
Протромботические состояния |
Экстракраниальная допплерография (сонных артерий) и эхокардиография. Коагулограмма: определение протеина S, протеина С, антитромбина III, выявление люпусного антикоагулянта, определение концентрации тканевого активатора плазминогена, активность ингибиторов активатора плазминогена |
Спиноцеребеллярные дегенерации |
Клиническое течение и отсутствие изменений других лабораторных тестов |
Митохондриальные энцефалопатии |
Определение лактата и пирувата в плазме и ЦСЖ, определение мутаций митохондриальной ДНК, биопсия скелетных мышц для выявления митохондриальных нарушений |
КАДАСИЛ (церебральная аутосомно-доминантная артериопатия с субкортикальными инфарктами и лейкоэнцефалопатией) |
Клиническое течение, анализ генетического сцепления, артериопатические проявления |
Таблица 3. Заболевания, сходные с РС по клиническим, МРТ- и ЦСЖ-критериям:
Заболевания |
Исследования для подтверждения диагноза |
Васкулиты: синдром Шегрена, узелковый полиартериит, системная красная волчанка |
Определение антител к кардиолипину, антинуклеарного фактора, волчаночного антикоагулянта, антител к нативной ДНК, Ro/SS-A и La/SS-B. При необходимости – церебральная ангиография, ретинальная флюороангиография, исследование с помощью щелевой лампы и биопсия |
Болезнь Бехчета |
Клинические проявления (язвенное поражение слизистых оболочек глаз, полости рта, кожи половых органов и нервной системы) |
Нейроборрелиоз |
Определение антител к Borrelia burgdorferi в сыворотке и ЦСЖ. Положительные реакции подтверждены Lues-TPHA-тестом |
Саркоидоз |
Тест Квейма, определение уровня ангиотензин-превращающего фермента в сыворотке и ЦСЖ, биопсия любого доступного очага (гранулемы, состоящие из эпителиоидных, гигантских клеток и макрофагов). |
Адренолейкодистрофия |
Определение длинноцепочечных (высших) жирных кислот |
Инфекция HTLV-I (тропический спинальный парапарез) |
Определение антител к HTLV-I |
Зрительная атрофия Лебера |
Анализ митохондриальной ДНК для исключения мутации в положении 11778 |
Заболевания, сходные с рассеянным склерозом по клиническим критериям, но имеющие четкие отличия на МРТ:
• Гранулематоз Вегенера – гранулематоз глазницы, придаточных пазух носа, мультифокальное поражение белого вещества больших полушарий;
• Болезнь Уиппла (Whipple’s) – множественные очаговые изменения, церебральная атрофия, расширение желудочков;
• Мальформация Арнольда-Киари – опущение миндалин мозжечка, продолговатого мозга, четвертого желудочка через большое затылочное отверстие в позвоночный канал, гипоплазия мозжечка, энцефалоцеле в затылочной области с внедрением мозжечка в грыжевой мешок;
• Экстра- и интрамедуллярные компрессионные поражения – метастатическая опухоль, лимфома, миеломная болезнь, эпидуральные абсцесс, гематома, туберкулезный спондилит, первичные опухоли спинного мозга, подвывих в атлантоаксиальном сочленении;
• Внутричерепное новообразование – опухоль, перифокальный отек, смещение срединных структур, сдавление желудочков, гидроцефалия.
20. Принципы лечения РС
Лечение. Лечение обострений проводят кортикостероидами ( 1-2 г метилпреднизолона в течение 3-7 дней («пульс-терапия»))
В конце курса целесообразны инъекции АКТГ. При тяжелых обострениях наряду с кортикастероидами проводят плазмаферез, гемосорбцию.При наличии каких-либо общеинфекционных признаков проводятся курсы антибиотикотерапии. Для предупреждения обострений при ремиттирующем течении заболевания длительно проводят лечение иммуномодулирующими средствами: бета-интерферон (бетаферон) или авонекс, ребиф, или глатирамера ацетат (копаксон).
Показаны препараты, влияющие на тканевый обмен: аминокислоты (шутаминовая кислота, глютаминат кальция, метионин, кортексин, актовегин), аминалон, витамины группы В, пирацетам, ноотропил, АТФ, ко-карбоксилаза, никотиновая кислота или ее препараты. Используют гипербарическую оксигенацию, обменный плазмаферез. В связи с изменением реологических и свертывающих свойств крови показаны антиагрегатные препараты: пентоксифиллин (трентал), курантил, гепарин, корректоры микроцирк
21. Патогенетическая терапия РС

22. Симптоматическая терапия РС
23. Профилактика осложнений при РС
24. Общая характеристика острого рассеянного энцефаломиелита
Острый рассеянный энцефаломиелит (ОРЭМ) — патология нервной системы, которая возникает вследствие аутоиммунных нарушений. При ОРЭМ происходит повреждение миелиновой оболочки, которая находится вокруг нервных волокон, в результате нарушается работа головного и спинного мозга. 2
Некоторые характеристики заболевания:
Начало острое, схоже с появлением респираторной инфекции: повышается температура тела, возникает озноб, слабость. 2
Признаки неврологических нарушений возникают спустя несколько дней после первого эпизода повышения температуры: спутанность сознания, нарушение координации, судороги, эпилептические припадки, речевые расстройства, нарушение глотания, сонливость, галлюцинации, нарушение дыхания. 2
При поражении глазного нерва возникают зрительные нарушения: двоение в глазах, белые пятна перед глазами, сложность при фокусировке. 2
Нарушения сознания могут приводить к коматозному состоянию. 2
Заболевание редкое, чаще всего фиксируется в детском возрасте, что связано с особенностями иммунной системы. 2
Хорошо поддаётся лечению при своевременно начатой терапии, в большинстве случаев заболевание не оставляет последствий. 2
При первых признаках поражения нервной системы следует обратиться за медицинской помощью.
25. Критерии диагностики ОРЭМ

Этиология и патогенез. Чаще всего заболевание азвивается на фоне перенесенной респираторной инфекции или на фоне гнойного синусита, отита. Точного возбудителя идентифицировать пока не удалось. Однако в патогенезе болезни ведущую роль играют аллергические реакции, приводящие к демиелинизирующему процессу в головном и спинном мозге.
Основу патаморфологического процесса составляют периваскулярные множественные очаги демиелинизации с участием микроглии. Локализация процесса различна:
белое вещество больших полущарий мозга,
ствол мозга,
спинной мозг.
Периаксиальный демиелинизирующий процесс обнаруживается и в спинномозговых корешках и нервах конечностей.
26. Лечение ОРЭМ
Лечение. Симптоматическое. В острый период показаны десенсибилизирующие,
дегидратирующие препараты. Назначают АКТГ (до 80 ЕД в день) или кортикостероиды, иммуноглобулины. При выраженных бульбарных нарушениях проводят реанимационные мероприятия. Используют глиатилин, дибазол в малых дозах, витамины группы В, ноотропные, нейропротекторные препараты.
27. Дифференциальная диагностика ОРЭМ
Диагностика. Диагноз основывается на остром инфекционном начале, наличии
симптомов мультифокального полисистемного поражения. Дифференцировать
рассеянный энцефаломиелит следует от:
энцефаломиелитов при кори,
ветряной оспе,
краснухе.
Основное значение при этом имеют данные анамнеза. Более сложен дифференциальный диагноз с рассеянным склерозом.
В большинстве случаев окончательный диагноз устанавливается после
длительного наблюдения . Наличие рецидивов и ремиссий свидетельствует о рассеянном склерозе.
28. Общая характеристика болезни Шильдера
Болезнь Шильдера (лейкоэнцефалит Шильдера) — редкое заболевание, характеризующееся массивной деструкцией миелина в белом веществе головного мозга. Преимущественно встречается у детей и подростков. 2
Общая характеристика болезни Шильдера:
Этиология до конца не изучена. Предполагается, что в основе патоморфологических изменений лежит процесс демиелинизации белого вещества больших полушарий головного мозга, при этом осевые цилиндры поражаются в меньшей степени. 2
Характерным признаком является крупный, чётко очерченный, асимметричный очаг разрушения миелина, который часто затрагивает целую долю или полушарие головного мозга. В некоторых случаях могут быть симметрично поражены оба полушария. 2
Клиническая картина включает тяжёлые неврологические симптомы, включая эпилептические припадки, центральные парезы, когнитивные нарушения, афазию, апраксию, а также нарушения зрения и слуха. 2
Заболевание часто имеет прогрессирующее течение, и смерть пациента, как правило, наступает в период от 6 месяцев до 3 лет с момента появления первых симптомов. 2
Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. 1
Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами, однако лечение малоэффективно.
29. Лечение болезни Шильдера
Специфического лечения болезни Шильдера не существует. Терапия в основном носит поддерживающий характер и направлена на облегчение симптомов. 14
Врач может назначить:
кортикостероиды; 4
бета-интерферон; 4
иммуносупрессивные препараты; 4
физиотерапию; 4
трудотерапию; 4
пищевую поддержку на более поздних стадиях. 1
Основой терапии является подавление иммунитета. 1
Прогноз болезни Шильдера непредсказуем. Чаще всего заболевание прогрессирует в течение жизни, приводя к инвалидизации. В ряде случаев болезнь становится фатальной, в других отмечается явное улучшение состояния вплоть до полной ремиссии.
30. Нейродегенеративные заболевания. Общая характеристика.
Нейродегенеративные заболевания — группа медленно прогрессирующих болезней нервной системы 1. Для них характерна гибель нейронов (нейродегенерация) 1.
Этот необратимый процесс приводит к клиническим проявлениям: деменции (ухудшение когнитивных способностей), нарушению двигательных функций, снижению чувствительности человека 1.
Примеры нейродегенеративных заболеваний: болезнь Альцгеймера, Паркинсона, хорея Хантингтона, мультисистемная атрофия, хроническая травматическая энцефалопатия и другие 1.
Причины гибели нейронов могут состоять в мутации гена (наследственная форма заболевания), воспалении и окислительном стрессе (приобретённая форма) 1. Триггером развития болезни может стать травма, инфекция, нарушения сосудов мозга
Нейродегенеративные заболевания приводят к постепенному нарастанию психической и физической беспомощности. Прогрессирующая патология головного мозга приводит к когнитивным и двигательным нарушениям. Но на начальных этапах все же сохраняется критика и самокритика, что влечёт переживания от осознания собственной несостоятельности.
31. Нейродегенеративные заболевания с поражением экстрапирамидной системы: болезнь Паркинсона, болезнь Гентингтона, болезньКоновалова-Вильсона.
Болезнь Паркинсона — медленно прогрессирующее дегенеративное заболевание центральной нервной системы, основными проявлениями которого являются такие двигательные нарушения, как гипокинезия, ригидность мышц, тремор покоя, постуральные расстройства. Кроме этого при болезни Паркинсона развиваются вегетативные, аффективные и другие расстройства.
Классификация болезни Паркинсона основывается на возрасте начала болезни:
ювенильная (ювенильный паркинсонизм)
с ранним началом
с поздним дебютом
Также известны различные классификации синдрома паркинсонизма:
дрожательные
дрожательно-ригидные
ригидно-дрожательные
акинетико-ригидные
смешанные
Этиология.Причина остается в значительной степени неизвестной. Предполагается, что на возникновение заболевания влияют генетические факторы, внешняя среда (возможное воздействие различных токсинов), процессы старения. Генетические факторы имеют доминирующее значение при раннем развитии болезни Паркинсона. Молодые пациенты с этим заболеванием и с семейной историей болезни с большей вероятностью переносят гены, связанные с болезнью Паркинсона, такие, как SNCA, PARK2, PINK1 и LRRK2.
Патогенез.Болезнь Паркинсона относится к группе синуклеинопатий, так как избыточное накопление в нейронах альфа-синуклеина приводит к их гибели. Повышенный уровень альфа-синуклеина может быть следствием нарушения внутриклеточной системы клиренса белков, осуществляемого лизосомамии и протеосомами. У пациентов обнаружено нарушение функционирования указанной системы, среди причин которого указывают старение, окислительный стресс, действие воспаления, токсины окружающей среды. Клетки гибнут предположительно из-за активации генетически запрограмированного механизма (апоптоза).
Клиника.Многие симптомы болезни Паркинсона не связаны с движением. Немоторные («невидимые симптомы») болезни Паркинсона распространены и могут влиять на повседневную жизнь больше, чем более очевидные трудности с движением. Они могут включать:
нарушение обоняния;
расстройства сна;
когнитивные симптомы (снижение памяти, легкомысленность);
запор;
расстройства мочеиспускания;
повышенное потоотделение;
сексуальную дисфункцию;
усталость;
боль (особенно в конечностях);
покалывание;
беспокойство и депрессию.
В начале заболевания нередко ставится неверный диагноз — плечелопаточный периартрит, проявляющийся болью и напряжением в мышцах руки и спины.
Синдром паркинсонизма является основным клиническим проявлением болезни Паркинсона, его симптомы:
замедленность всех движений;
истощаемость быстрых повторяющихся движений в руках и ногах;
скованность мышц (мышечная ригидность);
дрожание рук и ног (но почти никогда — головы), наиболее выражено в покое;
неустойчивость при ходьбе;
укорочение длины шага и шарканье при ходьбе, топтание на месте, застывания при ходьбе, отсутствие cодружественных движений руками при ходьбе.
Вначале симптомы возникают только с одной стороны тела, но постепенно приобретают двусторонний характер. Симптомы остаются выраженными на той стороне, где возникли в начале заболевания. Симптомы на другой стороне тела часто не становятся такими же тяжелыми, как симптомы на начальной стороне. Движения становятся все более замедленными (основной симптом паркинсонизма). Симптомы заболевания колеблются в течения дня и зависят от многих факторов.
В зависимости от преобладания в клинической картине того или иного симптома выделяют следующие формы:
1. Смешанная (акинетико-ригидная-дрожательная) форма характеризуется наличием всех трёх основных симптомов в разном соотношении.
2. Акинетико-ригидная форма характеризуется выраженными признаками гипокинезии и ригидности, к которым обычно рано присоединяются нарушения ходьбы и постуральная неустойчивость, при этом тремор покоя отсутствует или выражен минимально.
3. Дрожательная форма характеризуется доминированием в клинической картине тремора покоя, признаки гипокинезии уходят на второй план.
Различают пять стадий болезни Паркинсона, каждая из которых отражает степень тяжести заболевания. Наибольшее распространение получила классификация, предложенная в 1967 году Хеном и Яром:
0 стадия — двигательные проявления отсутствуют
I стадия — односторонние проявления заболевания
II стадия — двусторонние симптомы без постуральных нарушений
III стадия — умеренная постуральная неустойчивость, но пациент не нуждается в посторонней помощи
IV стадия — значительная утрата двигательной активности, но пациент в состоянии стоять и передвигаться без поддержки
V стадия — в отсутствие посторонней помощи пациент прикован к креслу или постели
Лечение.Продолжительность и качество жизни пациента непосредственно зависит от выбранной терапии и своевременно начала приема препаратов. Требуется постоянный контроль состояния здоровья лечащим неврологом.
На ранних стадиях используется медикаментозное лечение препаратами, активирующими выработку и синтез дофамина. Дополнительно применяются препараты, снижающие распад данного нейромедиатора.
Назначаются антагонисты рецепторов дофомина. Они выполнятся на основе прамипексола, ропинирола, ротиготина.
Назначаются МАО-ингибиторы типа Б, основанные на резалгине.
К третьей стадии начинается назначение Леводопы. У пациентов старше 70 лет использование данного средства рекомендовано с момента диагностирования.
Минусом Леводопы становится быстрое привыкание с необходимостью стабильного увеличения дозировки. В старшей возрастной группе Леводопа назначается с учетом сопутствующих возрастных диагнозов.
Дополнительно рекомендован прием витаминов, с включением магния. Витаминов А и D. В диету включается шпинат, морковь, помидоры, зеленый лук и другие овощи.
При отсутствии противопоказаний может быть рекомендовано оперативное вмешательство:
глубокая электростимуляция головного мозга;
стереотаксия;
использование стволовых клеток способных при подсадке заменять погибшие нейроны.
Операция часто заметно улучшает состояние пациента.
При лечении паркинсонизма используется два типа лекарственных средств:
противопаркинсонические антихолинергические лекарственные средства, к которым относится динезин, беллазон, циклодол, этпенал, тро-пацин, норакин;
противопаркинсоническиедофаминэргические препараты левопа, глудантан, мидантан, депренил.
Лекарства имеют противопоказания. Многие способны провоцировать отрицательные реакции. В том числе провоцировать галлюцинации.
Хорея Гентингтона — это наследственное, медленно прогрессирующее заболевание нервной системы, характеризующееся хореическими гиперкинезами, психическими нарушениями и прогрессирующей деменцией. Заболевание диагностируется с помощью молекулярно-генетического анализа и томографии головного мозга. Этиотропного лечения не разработано. Пациентам проводится симптоматическая терапия, направленная на подавление гиперкинезов. Прогноз неблагоприятный.
Этиология.Ген хореи Гентингтона находится на коротком плече хромосомы 4р16.3. Он кодирует белок гентингтин, функция которого до конца не выяснена. Однако известно, что аномальный белок токсичен для нейронов головного мозг, что и обусловливает постепенную атрофию церебральных структур. Способ наследования - аутосомно-доминантный, возможна передача дефектного гена как по женской, так и по мужской линии.
Патогенез.Хорея Гентингтона развивается в результате увеличения числа тринуклеотидных повторов — цитозин-аденин-гуанин, расположенных в первом экзоне гена. Триплет цитозин-аденин-гуанин кодирует аминокислоту глутамин, поэтому в белке образуется удлиненный полиглутаминовый тракт. Формируя подобие «замка-застежки», расширенный полиглутаминовый участок белка гентингтина изменяет свою собственную информацию и прочно соединяется с другими белками. В результате происходит агрегация белков, нарушаются межбелковые взаимодействия, что приводит к апоптозу клеток.
Клиника.Хорея Гентингтона манифестирует, как правило, в возрасте от 20 до 50. Случаи ювенильной формы заболевания довольно редки (не более 10%); наиболее ранний дебют заболевания, известный на сегодняшний день — 3 года. Типичное проявление хореи Гентингтона у взрослых — хореический синдром, в подростковом возрасте он встречается редко. Локализация хореических гиперкинезов приходится на лицевую мускулатуру, что вызывает выразительные гримасы с высовыванием языка, подергиванием щек, поочередным нахмуриванием и/или приподниманием бровей.
В ряде случаев наблюдались гиперкинезы в руках в виде быстрого сгибания и разгибания пальцев, в ногах — в виде поочередного скрещивания и разведения ног в сторону. Движения обычно не столь стремительны, как при малой хорее, но сложнее, иногда замедленные (по типу атетоидных). С прогрессированием хореи Гентингтона гиперкинезы усиливаются, приобретают характер атетоза и резко выраженной дистонии, впоследствии переходящей в ригидность.
При ювенильных формах хореи Гентингтона в 50% случаев заболевание манифестирует в виде брадикинезии и ригидности. Судороги возникают в 30-50% случаев (в отличие от взрослых пациентов). С развитием заболевания у больных происходит расстройство речевой функции. В первую очередь возникают проблемы со звукопроизношением, семантическая и синтаксическая структура речи остается сохранной до последней стадии заболевания. Со временем меняются скорость речи и ее ритм.
Глазодвигательные нарушения наблюдаются в большинстве случаев на ранних стадиях хореи Гентингтона. У пациентов нарушается автоматизация саккадирующих движений глазных яблок: удлиняется латентный период начала саккадирующих движений глаз, снижается скорость перевода взора и точность слежения. С развитием заболевания у большинства пациентов возникает вертикальный, реже горизонтальный, иногда комбинированный нистагм.
Прогноз.В большинстве случаев хореи Гентингтона прогноз на жизнь малоблагоприятный. Смерть, обусловленная различными осложнениями (пневмония, застойная сердечная деятельность), наступает через 10-13 лет течения заболевания. Продолжительность жизни пациентов колеблется от 45 до 55 лет.
Лечение носит преимущественно симптоматический характер.Согласно некоторым исследованиям, длительный прием коэнзима Q10, антиглутаматергических средств (например, мемантина), креатина, миноциклина (ингибитора каспаз с антиапоптозной активностью), витамина Е может несколько замедлять прогрессирование заболевания,
При умеренно выраженной хорее препаратом первого выбора может служить амантадин в дозе 200-500 мг/сут.
При его недостаточной эффективности к нему могут быть добавлен:
клоназепам (1—6 мг/сут.);
атипичные нейролептики клозапин (12,5—75 мг/сут.;
рисперидон (1—2 мг/сут.).
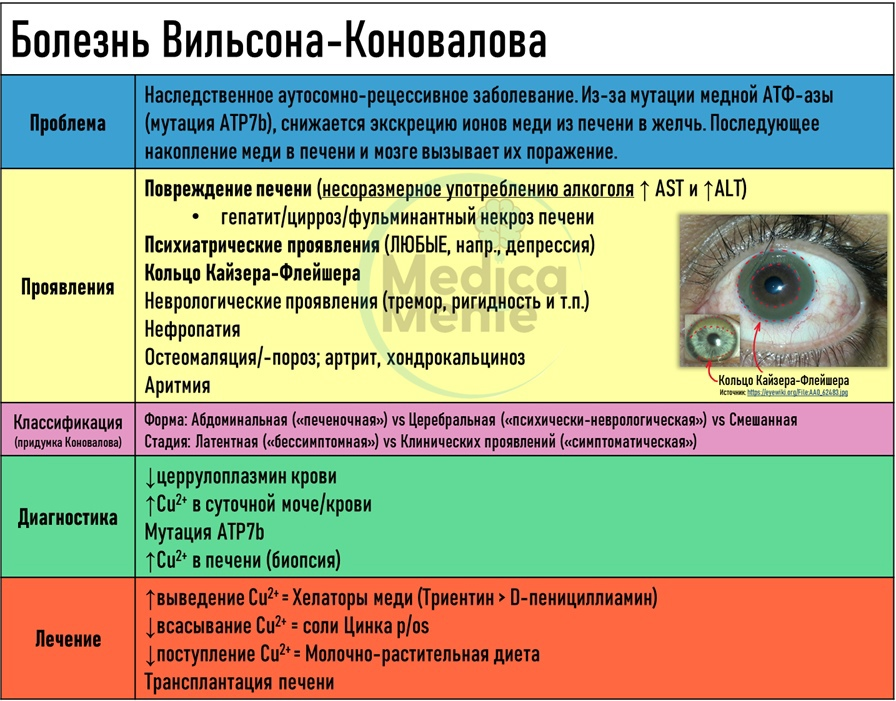
32. Нейродегенеративные поражения координаторной системы (ранние и поздние мозжечковые атаксии, атаксия Фридрейха); Ранняя мозжечковая атаксия может быть, например, представлена атаксией с селективным дефицитом витамина Е. Заболевание начинается чаще всего в возрасте от 4 до 18 лет и характеризуется постепенно прогрессирующими координаторными нарушениями, дизартрией, сухожильной арефлексией, нарушением суставно-мышечной и вибрационной чувствительности. 5
Поздняя мозжечковая атаксия может быть идиопатической, что означает, что дегенерация мозжечка происходит без очевидной причины. Такой тип атаксии обычно начинается примерно в 50-летнем возрасте.

пирамидной системы (болезнь Штрюмпеля);
болезнь Штрюмпеля (семейная спастическая параплегия) — наследственное хроническое заболевание, при котором происходит двустороннее поражение пирамидных трактов (системы нервных структур, отвечающих за координацию движений) в боковых и передних канатиках спинного мозга. 12
Некоторые симптомы болезни Штрюмпеля:
мышечная слабость (чаще в ногах), которая затрудняет перемещение, а на поздних стадиях заболевания делает его невозможным; 1
повышенный мышечный тонус в ногах; 1
истончение мышц; 1
скованность в ногах, в некоторых случаях — снижение чувствительности; 1
повышенная мышечная утомляемость; 1
укорочение стопы и увеличение её высоты; 1
в редких случаях могут появиться нарушение внятности речи и недержание мочи. 1
Причина болезни Штрюмпеля пока не определена. Чаще всего она развивается на генетическом уровне и может проявляться у пациентов любого возраста. 1
Полностью избавиться от болезни Штрюмпеля невозможно, но можно улучшить качество жизни пациента: назначаются препараты, расслабляющие мышцы, и ношение ортезов, помогающих передвигаться. 1
Для диагностики заболевания проводят МРТ спинного мозга, а при наличии противопоказаний к МРТ — КТ.
поражения спинного мозга(спинальные амиотрофии);
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. 2
Некоторые виды СМА:
СМА 0 типа. Симптомы начинают развиваться ещё внутриутробно и проявляются в первые дни жизни ребёнка. 3
СМА 1 типа. Развивается в течение первых 6 месяцев жизни ребёнка. Проявляется слабостью, отставанием в физическом развитии, проблемами с глотанием и жеванием. 3
СМА 2 типа. Стартует в возрасте от 3 месяцев до полутора лет. После старта СМА у ребёнка начинают выпадать двигательные функции, со временем он утрачивает способность ходить. 3
СМА 3 типа. Первые симптомы появляются от полутора до 19 лет. Атрофия начинается с бедренных мышц и постепенно распространяется на другие. Отличается медленным течением и более благоприятным прогнозом. 3
СМА 4 типа. Симптомы появляются во взрослом возрасте, часто после 30 лет. Проявления этого вида СМА схожи с боковым амиотрофическим склерозом. 3
Некоторые симптомы СМА: отсутствие рефлексов, общая мышечная слабость, вялость, низкий мышечный тонус, трудности в достижении основных этапов развития ребёнка, снижение тонуса дыхательных мышц и другие. 5
Диагностика осуществляется при помощи генетического тестирования. 4
Лечение носит поддерживающий характер. Оно включает в себя правильное питание, хороший уход, при необходимости респираторную поддержку, лечебную физкультуру, массажи, физиотерапию. 41
Вылечить СМА полностью невозможно, так как заболевание вызвано генетическими нарушениями. Прогноз зависит от формы СМА и возраста её дебюта
невральные поражения (наследственнаясенсомоторная полинейропатия).

33. Деменции. Общая характеристика. Корковые деменции. Болезнь
Альцгеймера.

Деменция — это хроническое, прогрессирующее и необратимое заболевание, которое характеризуется нарушениями когнитивных способностей. Оно связано с ухудшением памяти, способности к обучению и принятию решений. 1
Выделяют три стадии заболевания:
Начальная или лёгкая. Симптомы невыраженные: у пациентов может снижаться концентрация внимания, они быстро устают, становятся вялыми. 4
Умеренная. Клинические признаки болезни более выражены: у пациентов сильно нарушается память, ухудшается характер. 4
Тяжёлая. Пациенты на этом этапе полностью зависят от окружающих их людей: без посторонней помощи не способны выполнить даже простых действий, перестают контролировать мочеиспускание и дефекацию. 4
Деменция неизлечима, поэтому терапия носит скорее поддерживающий характер. Как правило, лечение включает в себя медикаментозные методы, психотерапию, физиолечение. 1


34. Подкорковые деменции. Когнитивные нарушения нейродинамического типа.



35. Деменция лобного типа. Болезнь Пика (фронто-темпоральная
деменция).

Клинические проявления могут быть очень разнообразными. Выделяют две основные формы болезни: поведенческую и речевую.
В целом у человека с болезнью Пика нарушается и речь, и поведение, но симптомы одной группы, как правило, преобладают над проявлениями другой. Однако в обоих случаях признаки болезни появляются и прогрессируют постепенно, а со временем человек перестаёт осознавать, что болен.
Поведенческая форма
Основные симптомы этой формы — выраженные нарушения поведения, которые проявляются в виде утраты манер или внешнего приличия, сексуальной несдержанности, социально неуместных, импульсивных, необдуманных или непредусмотрительных поступков, например человек может публично раздеваться, трогать или целовать незнакомых людей.
Также часто отмечаются волевые нарушения в виде апатии или нежелания что-либо делать. Пациенты могут стать неопрятными, пренебрегать личной гигиеной, разбрасывать мусор. Характерным признаком является утрата социальных навыков (человек становится замкнутым), эмпатии (нарушается способность к состраданию), снижается отзывчивость к чувствам и потребностям близких людей. Например, он перестаёт воспринимать чужие личные границы, учитывает только свои интересы и потребности, становится равнодушным к чужой боли или горю. Также могут возникать признаки ранее несвойственного ритуального и стереотипного поведения, например пациент может постоянно раскачиваться, сжимать губы, чмокать, похлопывать руками, топать ногами, растирать или пощипывать кожу, одежду и т. д.
Эта форма болезни Пика может также проявляться изменением пищевого поведения с появлением нетипичных ранее пищевых пристрастий, например переедания, повышенной тяги к алкоголю и курению, употреблением несъедобных предметов [4].
При всём этом сохраняется эпизодическая память и зрительно-пространственные функции: пациент может сам почистить зубы, полить цветы, знает, где находится кухня, и т. д. Благодаря этому он может оставаться самостоятельным в быту. Также обрывочно он помнит некоторые факты из профессиональной и личной жизни.
Примерно у половины пациентов с поведенческой формой болезни Пика через несколько лет от начала заболевания начинают появляться симптомы поражения двигательного нейрона, что проявляется нарушением артикуляции, глотания, парезами (снижением силы мышц) и, как следствие, нарушением ходьбы [18].
Речевая форма
Ведущим симптомом является выраженный дефект речи, который значимо влияет на повседневную активность пациента. При этом серьёзные поведенческие нарушения чаще всего отсутствуют. Основным симптомом будет афазия — нарушение речи разной выраженности.
Проявления речевой формы могут быть очень разнообразны. Иногда родственники пациента могут замечать за ним только снижение темпа речи, заикание, «запинки». Могут наблюдаться парафазии — замена близких по звучанию слов, например «лодка» — «ложка». В речи становится меньше прилагательных и наречий, человек начинает использовать более общие понятия ( «они», «вещи», «эти») вместо конкретных имён или названий предметов. Часто происходит замена слов в пределах тематики, так «лев» может стать «волком», а «телевизор» — «холодильником».
У таких пациентов речь становится сильно замедленной, монотонной, неэмоциональной, с ограниченным набором слов — так называемый «телеграфный стиль». Часто человек не может цокать, свистеть и облизывать губы языком.
При этом пациент может долгое время всё так же понимать слова и названия предметов. В конце заболевания формируется мутизм — полная утрата способности отвечать на вопросы и в целом разговаривать.
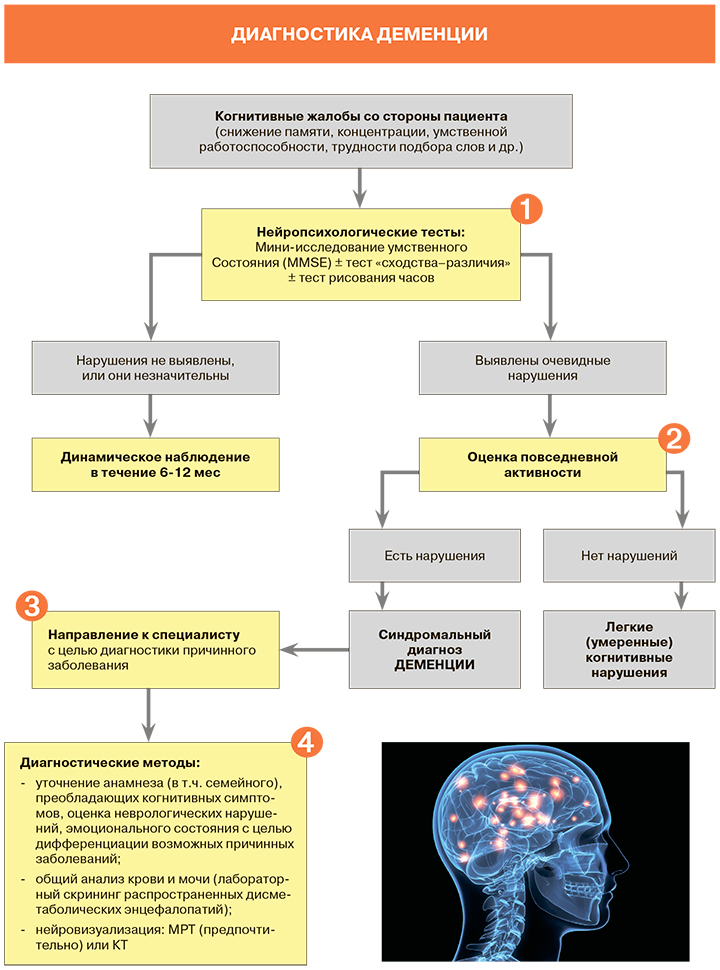
36. Диагностика деменции.
Поставить диагноз «деменция» может психотерапевт или психиатр. 2
Диагностика включает в себя ряд анализов и исследований:
Опрос пациента и его близких о самочувствии, проявлениях заболевания, любых изменениях настроения и поведения, примерной дате их появления, а также о наличии кровных родственников с деменцией. 1
Физический осмотр и назначение анализов крови для поиска других возможных причин симптомов, таких как недостаточная активность щитовидной железы, дефицит микроэлементов или витаминов Е и В12. 1
Психиатрическая оценка. Включает анализ поведения и настроения, памяти и мыслительных процессов, адекватности определения времени и пространства, качества принятия решений, навыков общения, зрительного контакта, речи. 1
Когнитивные и неврологические тесты. Дают возможность врачам оценивать память, мышление и языковые навыки человека. 1
Визуализирующие исследования. Позволяют изучать ткани мозга, изменения его структуры и функций. Используются для выявления опухолей, инсультов — нарушения кровообращения мозга, или других причин, способных вызвать слабоумие. 1
УЗИ сосудов шеи и головы. Безопасный способ оценки структуры, проходимости и деформации сосудов, выявления в них тромбов и бляшек, а также измерения скорости кровотока. 1
Единого
теста, с помощью которого можно выявить
раннюю стадию деменции, не существует.
Определение её типа — задача ещё более
сложная, поскольку симптомы и физические
изменения мозга при различных формах
заболевания часто совпадают.
37. Лечение деменции.
Вылечить деменцию полностью невозможно, терапия направлена на замедление прогрессирования заболевания и снижение выраженности его симптомов. 4
Лечение зависит от типа и степени тяжести деменции:
Деменция лёгкой степени. Назначают седативные препараты при излишней тревожности, антидепрессанты для улучшения эмоционального состояния. Также показаны средства, которые улучшают мозговую активность и передачу нервных импульсов. 1
Средняя степень старческой деменции. Назначают лекарственные средства для защиты нервных клеток, а также препараты для восстановления памяти и мышления. 1
Тяжёлая форма деменции. Применяют мощные лекарственные препараты, стимулирующие мозговую активность. Они принимаются на постоянной основе и позволяют надолго сохранить ясное сознание. При агрессивном поведении, галлюцинациях назначаются нейролептики. 1
Некоторые методы лечения:
Психотерапия. Пациента обучают упражнениям для тренировки памяти, развития логического мышления. 12
Арт-терапия. Помогает активизировать мозговую деятельность и выразить мысли и эмоции через рисунок, а не через слова. 2
Терапия воспоминаниями. Пациенты слушают любимую музыку, пересматривают фотографии, обсуждают события, которые произошли с ними в течение жизни, слушают аудиозаписи с голосами родных и друзей. 2
Также важна социальная поддержка, включающая помощь в бытовых вопросах, организацию режима дня и обеспечение безопасности пациента. 5
Важно поддерживать здоровый образ жизни: заниматься спортом, поддерживать мозг в тонусе, питаться правильно и часто проводить время с близкими. Регулярные посещения врача и поддержка близких помогают снизить негативные эффекты заболевания.
-1.Синдром полинейропатии.
Полинейропатия – это системная патология периферического отдела нервной системы, характеризующееся распространенным поражением периферических нервных волокон с развитием парезов, трофических и вегетососудистых нарушений, а также неправильным восприятием боли и температуры. Чаще всего симптомы присутствуют в дистальных отделах конечностей
Полиневропатии могут проявляться:
двигательными расстройствами (периферические тетрапарезы,
парапарезы, арефлексия, мышечная гипотрофия, у некоторых больных
наблюдаются фасцикуляции, крампи)
нарушением чувствительности (часто по типу «носков и перчаток»),
сенситивной атаксией (при поражении волокон глубокой
чувствительности), парастезиями, гиперэстезией, болью
вегетативной дисфункцией (вазомоторные нарушения, расстройства
потоотделения и др. проявления периферической вегетативной
недостаточности; в ряде случаев изменением АД, изменением со
стороны желудочно-кишечного тракта, нарушением мочеиспускания)
Большинство полиневропатий вовлекает все три типа волокон, что
проявляется комбинированной симптоматикой. В зависимости от характера
и глубины поражения нервных волокон симптомы могут быть как
позитивными («симптомы раздражения»), так и негативными («симптомы
выпадения»). Начальным проявлением полиневропатии часто служат
позитивные чувствительные симптомы (парастезия, боль, гиперэстезия),
нередко возникающие в дистальных отделах нижних конечностей, к
которым позднее могут присоединяться двигательные и вегетативные
симптомы. Важное диагностическое и прогностическое значение имеет
патогенетическая диагностика полиневропатии. Аксональные
полиневропатии обычно бывают следствием токсических и метаболических
поражений нервной системы. При этом поражаются как толстые
миелинизированные, так и тонкие немиелизированные или
слабомиелинизированные волокна. При демиелинизирующих
полиневропатиях первичной мишенью патологического процесса служат
шванновские клетки и их элементы, преимущественно страдают крупные
миелинизированные волокна (моторные и проводящие глубокую
чувствительность)
2. Классификации полинейропатий.
По этиологии:Воспалительные, Токсические, Аллергические, Травматические.
По патоморфологии повреждения: Аксональные, Демиелинизирующие.
По характеру течения: Острые, Подострые, Хронические.


3. Дифференциальная диагностика аксонопатий и миелинопатий
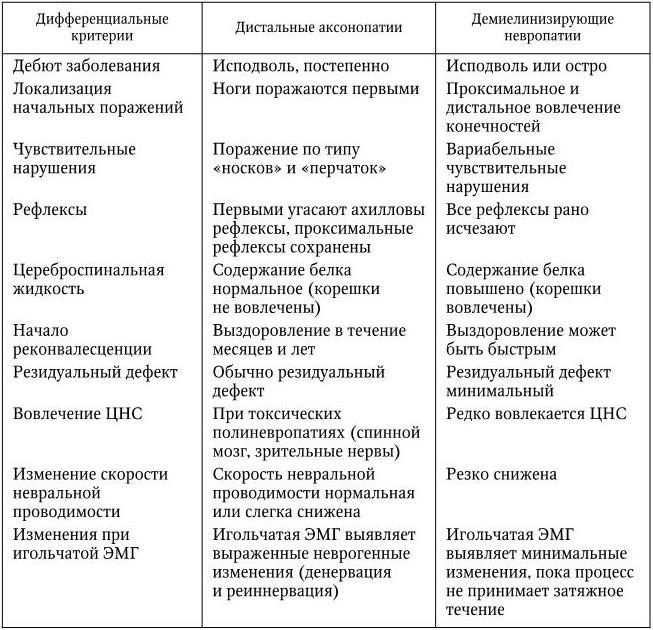
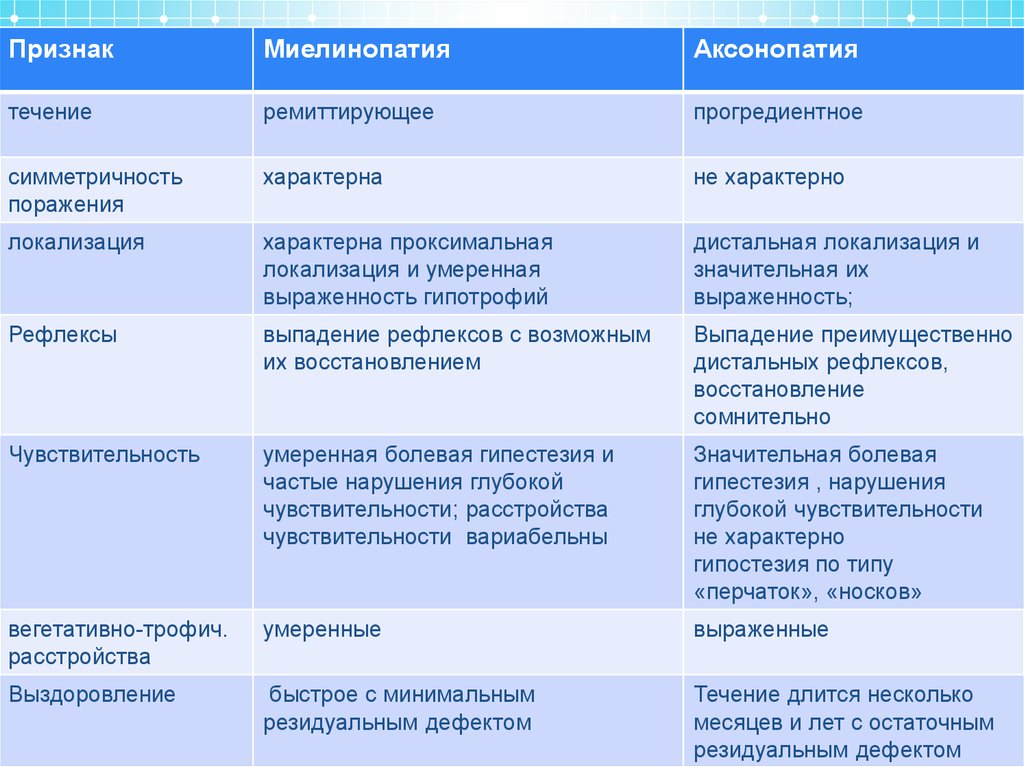
При аксонопатии: начало-постепенное; нижние конечности поражаются первыми; нарушение чувст-ти по типу носков и перчаток; первыми угасают ахилловы рефлексы; в цереброспинальной жидкости содержание белка в норме; выздоровление в течение месяцев или лет; скорость проведения понервам нормальная или слегка снижена. При миелинопатии: начало постепенное или острое; проксимальное и дистальное вовлечение конечностей; вариабельные нарушения чувст-ти; все рефлексы исчезают рано; в цереброспинальной жидкости повышенное содержание белка; выздоровление может быть быстрым; скорость проведения по нервам резко снижена.
Аксонопатия — это заболевание периферической нервной системы, при котором повреждения касаются аксонов (отростков нервных клеток). Это тяжёлые нарушения функций нервов, как следствие — атрофия мышц. 2
Причины аксонопатии:
сахарный диабет; 1
хроническая болезнь почек; 1
побочные эффекты химиотерапевтических препаратов (например, алкалоиды барвинка); 1
недостаточность питания (чаще всего недостатком тиамина или витамина B6, витамина B12 или витамина E); 1
злоупотребление алкоголем. 12
Симптомы аксонопатии:
вялый паралич мышц; 2
боль в конечностях; 2
изменение чувствительности кожи (может быть контраст ощущений на одном участке кожи по сравнению с другим); 2
нарушение речи; 2
чувство онемения лица, конечностей; 2
мышечная слабость конечностей; 2
нарушение координации движений; 2
сухость кожи; 2
очаговое побледнение, покраснение и посинение травмированного участка.
миелопатия — тяжёлая неврологическая патология, которая объединяет ряд острых и хронических заболеваний и синдромов, вызванных повреждением спинного мозга. 5
Некоторые виды миелопатии:
Спондилогенная — сдавление спинномозгового канала и ухудшение кровообращения, возникает при длительном остеохондрозе или из-за грыж межпозвоночных дисков. 1
Ишемическая (сосудистая, атеросклеротическая) — уменьшение поступления крови к позвоночнику. 1
Метаболическая — последствие эндокринологических нарушений (например, диабетическая). 1
Карциноматозная — метастатические повреждения костей, первичный онкологический процесс в лёгких, желудке, молочной железе, простате, поражение ЦНС из-за токсического воздействия рака. 1
Посттравматическая — последствия повреждений позвоночника, спинномозгового канала и кровеносных сосудов. 1
Инфекционная — возникает на фоне туберкулёзной инфекции, менингита, болезни Лайма, поражения золотистым стафилококком, ВИЧ-инфекции, нейросифилиса, из-за изменения структуры спинного мозга или образования абсцессов. 1
Радиационная — побочный эффект лучевой терапии во время лечения онкологического заболевания или случайного радиационного воздействия. 1
Димиелинизирующая — из-за разрушения миелиновой оболочки нервных волокон спинного мозга, часто встречается во время рассеянного склероза. 1
Некоторые общие симптомы миелопатии:
боль в спине в месте поражения; 1
уменьшение силы конечностей, снижение их функциональной активности: нарушение походки, трудности при письме или одевании; 1
нарушение всех видов чувствительности в руках и ногах; 1
расстройства мочеиспускания и дефекации.
4. Синдромология, ликворные изменения, лечение полинейропатии Гийена - Барре.
Острая воспалительная полирадикуломиелоневропатияаутаиммунной природы
характеризуется острой демиелинизацией корешков спинномозговых и черепных нервов, проявляется прогрессирующей мышечной слабостью с расстройствами
дыхания, отсутствием глубоких рефлексов.
Реже встречается аксональный вариант заболевания, который обычно протекает более тяжело.
Перенесенная инфекция служит провоцирующим фактором, запускающим аутаиммунную реакцию. Наблюдали этот синдром и при клещевом боррелиозе, БИЧ-инфекции, саркоидозе, системной красной волчанке, злокачественных новообразованиях. Заболевание рассматривается как аутоиммунное с деструкцией ткани, вторичной по отношению к клеточным иммунным реакциям. Обнаруживаются воспалительные инфильтраты в периферических нервах, а также корешках, сочетающиеся с сегментарной демиелинизацией.
Клиническая картина. Заболевание начинается с парестезий и болей в дистальных
отделах рук и ног, а иногда вокруг рта и в языке. Тяжелые нарушения чувствительности возникают редко. Вскоре присоединяется общая слабость, повышение температуры тела до субфебрильных цифр.
Иногда боли носят опоясывающий характер на туловище. Главным отличительным
признаком болезни служит мышечная слабость в конечностях, реже возникает слабость лицевых и бульбарных мышц. Двигательные нарушения раньше возникают
в ногах, а затем распространяются на руки.
Возможны поражения преимущественно проксимальных отделов конечностей;
при этом возникает симптомокомплекс, напоминающий миопатию. Нервные стволы болезненны при пальпации. Могут быть симптомы натяжения (Ласега, Нери).
Особенно выражены вегетативные нарушения - похолодание и зябкость дистальных отделов конечностей, акроцианоз, явления гипергидроза, иногда имеется гиперкератоз подошв, ломкость ногтей.
Типична белково-клеточная диссоциация в цереброспинальной жидкости. Уровень белка достигает 3-5 г/л. Высокие цифры белка определяются как при люмбальной, так и окципитальной пункции. Этот критерий очень важен для отличия синдрома Гийена-Барре-Штроля от спинальной опухоли, при которой высокие цифры белка обнаруживаются только при люмбальной пункции. Цитоз - не более 10 мононуклеарных клеток в 1 мкл.
Заболевание обычно развивается в течение 2-4 нед., затем наступает стадия стабилизации, а после этого - улучшение. Кроме острых форм, могут встречаться подострые и хронические. В подавляющем большинстве случаев исход заболевания благоприятный, но наблюдаются также формы, протекающие по типу восходящего паралича Ландри с распространением параличей на мышцы туловища, включая дыхательные, рук и бульбарную мускулатуру, что может приводить к смерти.
Диагностика.В анамнезе примерно у двух третей пациентов с острой воспалительной демиелинизирующей полинейропатией за 2 недели до появления мышечной слабости отмечается перенесение эпизода острой респираторной инфекции или гастроэнтерита, иногда – травмы или оперативное вмешательство, реже – переохлаждение или профилактическая вакцинация.
При физикальном обследовании следует обратить внимание на следующие особенности:
- ясное сознание;
- наличие мышечной слабости в ногах и/или руках;
- снижение или отсутствие сухожильных рефлексов, особенно в ногах;
- чувствительные нарушения по полиневритическому типу;
- наличие болей в конечностях, парестезий и дизестезий;
- относительная симметричность поражения;
- черепные нервы – особенно характерно поражение лицевого нерва, в 10-20%;
- вегетативные нарушения – высока вероятность развития тахикардии, аритмии, постуральной гипотензии, артериальной гипертензии, вазомоторных симптомов;
- симптомы прогрессируют в течение нескольких дней или недель, но нарастание симптоматики должно прекратиться к концу 4-й недели от дебюта болезни
Рекомендовано проведение люмбальной пункции с исследованием цереброспинальной жидкости.Характерным является повышение содержания белка в ликворе по прошествии 1 недели после появления характерных симптомов болезни;
присутствие в 1 мкл спинномозговой жидкости не более 50 моноцитов и/или 2 гранулоцитов; наличие феномена белково-клеточной диссоциации.
Рекомендовано проведение электронейромиографического исследования; характерно наличие электромиографических признаков демиелинизации и/или аксонального поражения периферических нервов.
Лечение.Специфической лекарственной терапии пока не существует.
Активным методом терапии является плазмаферез.
У больных частично удаляют плазму крови, возвращая форменные элементы.
Вводят человеческий иммуноглобулин внутривенно (0,4 г/кг ежедневно, 5 дней).
Применяются глюкокортикоиды (преднизолон по 1 -2 мгjкгjсут.), антигистаминные средства (димедрол, супрастин), витамины группы В, прозерин по 1 мл 0,05% раствора подкожно.
Важное значение имеет уход за больным с тщательным контролем за состоянием дыхательной и сердечно-сосудистой систем.
Дыхательная недостаточность в тяжелых случаях может развиваться очень быстро и приводит к смерти. Если у больного жизненная емкость легких оказывается менее 50% предполагаемого дыхательного объема, рекомендуется интубация или трахеостомия для проведения ИВЛ.
Выраженную артериальную гипертонию и тахикардию купируют применением антагонистов кальция (коринфар) и β-адреноблокаторов (пропранолол) в соответствующих состоянию больного дозах. Артериальную гипотонию лечат путем внутривенного введения жидкости с целью увеличения внутрисосудистого объема.
Необходимо каждые 1-2 ч осторожно менять положение больного в постели. Острая задержка мочевыделения и расширение мочевого пузыря могут вызвать рефлекторные нарушения, приводящие к колебаниям артериального давления и пульса. В таких случаях рекомендуется применение постоянного катетера.
В восстановительном периоде назначают ЛФК для предупреждения контрактур, массаж, озокерит, парафин, четырехкамерные ванны.
У большинства пациентов заболевание начинается с двигательных и/или чувствительных нарушений в ногах и/или руках, реже - с мышечных болей различной локализации. В ряде случаев боль может одновременно появляться вместе с онемением, парестезиями или двигательными нарушениями. Иногда первыми симптомами заболевания бывают нарушения глотания, изолированное двоение. Онемение, парестезии и слабость первоначально появляются преимущественно в нижних конечностях (до половины всех случаев) и распространяются спустя несколько часов или дней на верхние. Характерно поражение мимической мускулатуры, менее часты нарушения глотания. Нарушения тазовых функций в виде задержки мочеиспускания редки и наблюдаются в основном при тяжелых формах заболевания. Обычно они проходят через 3-5 дней после появления. Отмечаются также внезапные падения артериального давления и аритмии. Характерен гипергидроз туловища, ладоней, стоп. Состав спинномозговой жидкости при СГБ в первые дни болезни нередко нормален, но ее белок имеет четкую тенденцию к повышению после первых 5-7 суток, обычно достигая максимума к 3-4-й неделе. В ряде случаев не исключено резкое увеличение концентрации белка в ликворе уже спустя 2-3 суток после начала СГБ. Более чем 50 мононуклеарных лейкоцитов в ликворе или наличие в нем полиморфно-ядерных лейкоцитов исключают диагноз "СГБ". Лечебные мероприятия, проводимые при СГБ, подразделяют на специфические и неспецифические. Основными специфическими методами лечения заболевания в настоящее время являются программный плазмаферез и внутривенная пульс-терапия иммуноглобулинами класса G. К неспецифическим методам лечения относятся мероприятия, направленные на особый уход за больным и купирование разного рода осложнений, связанных с основным заболеванием, среди которых важнейшее место занимают лечение дыхательных и бульбарных нарушений. При этом дыхательная реанимация является, по существу, самостоятельным видом лечения при тяжелых формах СГБ. Только правильное сочетание специфических и неспецифических методов лечения способно в сжатые сроки привести к восстановлению большинство больных с тяжелыми формами СГБ.
4. Синдромология алкогольной полинейропатии
Наблюдается у лиц злоупотребляющих спиртными напитками, развивается в поздних стадиях заболевания. В патогенезе основная роль принадлежит токсическому действию алкоголя на нервы и нарушению в них обменных процессов. Изменения развиваются не только в спинальных и черепных нервах, но и в других отделах нервной системы (головном и спинном мозге). Появляются парестезии в дистальных отделах конечностей, болезненность в икроножных мышцах. Боли усиливаются при сдавлении мышц и надавливании на нервные стволы (один из ранних характерных симптомов). Вслед за этим развиваются слабость и параличи всех конечностей, более выраженные в ногах. Преимущественно поражаются разгибатели стопы. В паретичных мышцах быстро развиваются атрофии. Сухожильные и периостальные рефлексы в начале заболевания могут быть повышенными, а зоны их расширены. При выраженной клинической картине имеется мышечная гипотония с резким снижением мышечно-суставного чувства. Возникает расстройство поверхностной чувствительности по типу "перчаток" и "носков". В ряде случаев алкогольная полинейропатия может развиваться остро, чаще после значительного переохлаждения. Возможны при этом и психические расстройства. Могут наблюдаться вазомотроные, трофические и секреторные расстройства в виде гипергидроза, отёков дистальных отделов конечностей, нарушений их нормальной окраски и температуры. Из черепных нервов могут поражаться глазодвигательный, зрительный, реже вовлекаются в процесс блуждающий (ускорение пульса, нарушение дыхания) и диафрагмальный нервы.
Алкогольная полиневропатия – множественное поражение периферических нервов при алкоголизме. Обычно возникает на поздних стадиях алкогольной болезни. Сопровождается слабостью мышц, нарушениями чувствительности и атаксией. Возможна повышенная потливость. Нередко наблюдаются отеки, изменения температуры и окраски дистальных отделов конечностей. В ряде случаев возникают психические расстройства. Развивается постепенно, реже остро.
Клиника.Алкогольная полиневропатия чаще развивается подостро.
парестезии в дистальных отделах конечностейболезненность в икроножных мышцах, причем боли усиливаются при сдавлении мышц и надавливании на нервные стволы.
Вслед за этим - слабость и параличи всех конечностей, более выраженные в ногах. Преимущественно поражаются разгибатели стопы.
Атрофии в паретичных мышцах. Глубокие рефлексы нередко оказываются повышенными, а зоны их расширены. Однако при выраженной клинической картине имеется мышечная гипотония с резким снижением мышечно-суставногочувства.Расстройство поверхностной чувствительности по типу перчаток и носков.
Расстройства глубокой чувствительности приводят к атактическим нарушениям
В сочетании с выпадением глубоких рефлексов клиническая картина напоминает сифилитическую сухотку спинного мозга и даже получила название псевдотабеса. Однако при этом отсутствуют характерные для сухотки:
расстройства мочеиспускания,
боли по типу «прострела»,
положительная реакция Вассермана в ликворе и крови,
изменения зрачков.
Алкогольная полиневропатия может развиваться остро, чаще после значительного переохлаждения. Возникают изменения высших мозговых функций:вазомоторные, трофические и секреторные расстройства в виде гипергидроза,отеков дистальных отделов конечностей, нарушений их нормальной окраски и температуры.
Стадия нарастания болезненных явлений обычно продолжается недели и даже месяцы.
Затем наступает стационарная стадия и при лечении - стадия обратного развития. В общей сложности заболевание продолжается от нескольких месяцев до нескольких лет. При исключении употребления алкоголя прогноз обычно благоприятный.
Лечение.Назначают витамины группы В, С, ноотропные препараты.
В восстановительном периоде - прозерин, дибазол, физиотерапию.
5. Синдромология дифтерийной полинейропатии
Через 1-2 недели после начала заболевания могут возникнуть признаки поражения черепных нервов бульбарной группы: парез мягкого нёба, языка, расстройство фонации, глотания; возможно нарушения дыхания, особенно при вовлечении в процесс диафрагмального нерва. Поражение блуждающего нерва может обусловить бради- и тахикардию, аритмию. Нередко вовлекаются в процесс глазодвигательные нервы, что проявляется расстройством аккомодации. Реже наблюдается парез наружных глазных мышц, иннервируемых III, IV и VI черепными нервами. Полинейропатия в конечностях обычно проявляется поздними (на 3-4-ой неделе) вялыми парезами с расстройством поверхностной и глубокой чувствительности, что приводит к сенситивной атаксии. Иногда единственным проявлением поздней дифтерийной полинейропатии является выпадение сухожильных рефлексов.
6. Патогенез, синдромология, классификация, лечение
диабетической полинейропатии.
Развивается у лиц, страдающих диабетом. Полиневропатия может быть первым
проявлением диабета или возникает через много лет после его начала. Синдром полиневропатии встречается почти у половины больных диабетом.
Наиболее существенными механизмами в развитии невропатии являются ише- ·
мия и метаболические нарушения в нерве.
Ранним проявлением полиневропатии нередко может быть укорочение вибрационной чувствительности на лодыжках голеней и снижение ахилловых рефлексов. Эти
явления могут существовать многие годы.
Затем отмечается прогрессирование болезни: боли в стопах и голенях нарастают до
мучительных, боль часто обостряется под влиянием тепла и в покое. Выявляется слабость стоп. Нередко нарушается вегетативная иннервация. Если процесс прогрессирует, боли нарастают, появляются участки кожи, окрашенные в фиолетовый и черный цвет, мумификация гангренизированной ткани (диабетическая стопа). Часто в таких случаях возникает зуд.
Клиническая картина.Наиболее характерна периферическая нейропатия:больных беспокоят:
онемение в ногах
чувство ползания мурашек
судороги в конечностях
боли в ногах,усиливающиеся в покое,ночью и уменьшающиеся при ходьбе
Отмечается снижение или полное отсутствие коленных и ахилловых рефлексов,снижение тактильной и болевой чувствительности.
Иногда развиваетсяатрофия мышц в проксимальных отделах ног.
Возникают расстройства функции мочевого пузыря,у мужчин нарушается потенция.
Клинические варианты:
Симметричная дистальная сенсорная полинейропатия
Полинейропатия тонких волос
Симметричная проксимальная моторная нейропатия
Краниальная нейропатия
Множественная мононейропатия
Торакоабдоминальная мононейропатия
Ассиметричная проксимальная моторная нейропатия (диабетическая поясничнокрестцоваярадикулоплексопатия)
Диагностика.Зависит от формы диабетической нейропатии. На первичной консультации тщательно анализируются анамнез и жалобы на изменения со стороны сердечно-сосудистой, пищеварительной, дыхательной, мочеполовой, зрительной систем. У пациентов с диабетической нейропатией необходимо определение уровня:
глюкозы,
инсулина,
С-пептида,
гликозилированного гемоглобина крови;
исследование пульсации на периферических артериях,
измерение АД;
проведение осмотра нижних конечностей на предмет наличия деформаций, грибковых поражений, натоптышей и мозолей.
В зависимости от проявлений в диагностике диабетической нейропатии, кроме эндокринолога и диабетолога, могут участвовать другие специалисты:
кардиолог,
гастроэнтеролог,
невролог,
офтальмолог,
подолог.
Первичное обследование сердечно-сосудистой системы заключается в проведении ЭКГ, кардиоваскулярных тестов (пробы Вальсальвы, ортостатической пробы и др.), ЭхоКГ; определении уровня холестерина и липопротеидов.
Неврологическое обследование по поводу диабетической нейропатии включает проведение электрофизиологических исследований: электромиографии, электронейрографии, вызванных потенциалов.
Производится оценка рефлексов и различных видов сенсорной чувствительности:
тактильной с использованием монофиламента;
вибрационной - с помощью камертона;
температурной - путем прикосновения холодного или теплого предмета;
болевой - методом покалывания кожи тупой стороной иглы;
проприоцептивной – с помощью пробы на устойчивость в позе Ромберга.
К биопсии икроножного нерва и биопсии кожи прибегают при атипичных формах диабетической нейропатии.
Гастроэнтерологическое обследование при диабетической нейропатии предполагает проведение:
УЗИ органов брюшной полости,
ЭГДС,
рентгенографии желудка,
исследования пассажа бария по тонкой кишке,
тестов на хеликобактер.
При жалобах со стороны мочевыводящей системы исследуется:
общий анализ мочи,
выполняется УЗИ почек,
мочевого пузыря (в т. ч. УЗИ с определением остаточной мочи),
цистоскопия,
внутривенная урография,
электромиография мышц мочевого пузыря
Лечение.Эффективная терапия диабета, что важно для начальных проявлений диабетической полиневропатии. В остальном лечение проводится по общим принцилам реабилитирующей терапии.
Нормализация уровня глюкозы крови
Нормализация массы тела
Коррекция липидов крови
Обезболивание
Витамины группы В (В1 150-300 мг\сут)
Пентоксифиллин
α−липоевая кислота

7. Патогенез, клиника, дагностика, лечение полинейропатиий приуремии, болезнях печени, крови, системных болезняхсоединительной ткани, порфирии, лекарственных интоксикациях.
ВАЩЕ ХЗ ИНТЕРНЕТ В Помощь
8. Синдромология диагностика, лечение хронической воспалительной демиелинизирующей полинейропатии.
Клиническая классификация ХВДП
Варианты |
Характеристика |
Типичная ХВДП |
· симметричные проксимальная и дистальная слабость и чувствительные нарушения во всех конечностях, развившиеся в течение более 2 мес., сопровождающиеся отсутствием или снижением сухожильных рефлексов во всех конечностях, возможно поражение черепных нервов. Течение хронически прогрессирующее, ступенеобразно прогрессирующее, рецидивирующе-ремиттирующее. · подразумевает выделение одного из перечисленных ниже вариантов, остальные характеристики (период нарастания симптоматики, течение) соответствуют таковым типичной ХВДП (сухожильные рефлексы в непораженных конечностях могут быть нормальными): преимущественно дистальный вариант: сенсорные и двигательные нарушения локализуются только в дистальных отделах конечностей (дистальная приобретенная симметричная демиелинизирующая полиневропатия) |
Атипичная ХВДП |
· асимметричный вариант: сенсорные и двигательные нарушения асимметричны или локализуются в области иннервации отдельных нервов (мультифокальная приобретенная демиелинизирующая сенсорная и моторная невропатия, синдром Льюиса — Самнера); · фокальный вариант: сенсорные и двигательные нарушения локализуются в области иннервации плечевого или пояснично-крестцового сплетения, или одного и более периферических нервов в одной верхней или нижней конечности); · изолированный двигательный вариант: исключительно двигательные нарушения; · изолированный сенсорный вариант (включая хроническую иммунную сенсорную полирадикулопатию): исключительно чувствительные нарушения. |
ХВДП имеет четыре основных варианта течения [3,4]: · хронический монофазный - симптомы постепенно достигают максимальной выраженности, а затем подвергаются полному или частичному регрессу, в дальнейшем заболевание не прогрессирует и не рецидивирует; · хронический рецидивирующе-ремиттирующий - четко очерченные эпизоды усиления симптоматики с последующим обратным развитием — рецидивы, сменяются периодами стабилизации состояния, во время которых заболевание не прогрессирует — ремиссия; · ступенчато- прогрессирующий - прогрессирующее ступенеобразное нарастание симптомов; · неуклонно- прогрессирующий - медленное непрерывное нарастание симптомов.