

невра файл к эукзамену
.pdfЛечение:
Промывание желудка и клизмы для удаления остатков токсина.
Вводится антиботулиническая сыворотка (тип А, С, Е по 10 тыс. ME, тип В по
5000 ME).
Парентерально вводятся растворы для дезинтоксикации, коррекции кислотнощелочного равновесия и белкового баланса.
При необходимости осуществляется искусственная вентиляция легких, зондовое кормление.
Полное восстановление у выживших наблюдается в течение нескольких недель.
ПРОГРЕССИРУЮЩИЕ МЫШЕЧНЫЕ ДИСТРОФИИ
Прогрессирующие мышечные дистрофии представляют достаточно большую группу наследственных заболеваний, проявляющихся постепенно нарастающей мышечной слабостью и атрофией мышц. Они вызываются первичным наследственно обусловленным поражением мышц, при этом сохранными остаются двигательные нейроны, их аксоны и нервно-мышечные синапсы.
МИОДИСТРОФИЯ ДЮШЕНА
Этиология и патогенез:
При миодистрофии Дюшенна патологический ген локализован в коротком плече Х- хромосомы, при этом не происходит образования важного для скелетных мышц белка дистрофина, что вызывает некроз, фиброз и нарушение функции мышечных волокон. Заболевание передается по рецессивному, сцепленному с Х-хромосомой типу, поэтому болеют мальчики; их мамы могут быть бессимптомными носителями патологического гена, хотя при тщательном обследовании у них можно определить легкое снижение мышечной силы, увеличение размеров икроножных мышц и повышение содержания фермента креатинфосфокиназы в сыворотке крови.
Клиническая картина:
Заболевание проявляется уже в раннем детском возрасте отставанием в физическом развитии:
Больные дети позднее начинают ходить, часто плохо бегают и прыгают.
Мышечная слабость преобладает в проксимальных отделах ног и тазовом поясе
Изменения походки обычно обнаруживаются в возрасте 2—5 лет.
Икроножные мышцы, иногда четырехглавые мышцы бедра и дельтовидные увеличены в размере
181

и уплотнены за счет разрастания соединительной ткани (псевдогипергрофия),
сила их снижена.
Мышцы бедер, тазового и плечевого пояса атрофируются на ранних стадиях заболевания, усиливается поясничный лордоз.
Постепенно процесс принимает восходящее направление и распространяется на плечевой пояс, мышцы спины, а затем и на проксималные отделы рук. Чтобы встать, больной ребенок вынужден выполнять специальные приемы.
Часто страдает сердечная мышца по типу кардиомиопатии.
У многих больных снижен интеллект.
К 10 годам дети уже ходят с трудом, а к 15 годам они, как правило, полностью обездвижены. Больные обычно умирают на третьем десятилетии жизни вследствие сердечной недостаточности или легочных осложнений.
МИОДИСТРОФИЯ БЕККЕРА
Этиология: форма Х-сцепленной дистрофии, которая встречается реже (примерно в 10 раз) и протекает легче, чем миодистрофия Дюшенна. При ней белок дистрофин образуется, но количество его уменьшено, а структура изменена.
Клиническая картина:
Поражаются те же мышечные группы, что и при миодистрофии Дюшенна, но заболевание начинается в более позднем возрасте (от 5 до 45 лет),
Обычно заболевание не сопровождается кардиальными и интеллектуальными нарушениями, имеет лучший прогноз в отношении инвалидности и продолжительности жизни.
ЛИЦЕЛОПАТОЧНО-ПЛЕЧЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Этиология: наследуется по аутосомно-доминантному типу, патологический ген расположен в локусе 4q.
Клиническая картина:
Симптомы возникают в старшем детском или подростковом возрасте, реже у молодых людей.
Больным трудно поднимать руки над головой, наблюдаются крыловидные лопатки, слабость мимических мышц.
Больному не удается крепко зажмурить глаза и сжать губы
Атрофия и слабость наблюдаются преимущественно в мышцах плечевого пояса (трапециевидных, грудные грудино-ключично-сосцевидных, зубчатых, ромбовидных) и проксимальны» мышцах рук. З
аболевание прогрессирует медленно, многие пациенты достигают пожилого возраста. Сердечная функция и интеллект не нарушены.
182

Диагностика:
Анализ крови: повышение КФК;
Электромиографии — снижение длительности и амплитуды потенциалов двигательных единиц, отсутствие признаков поражения периферических нервов или мотонейронов передних рогов спинного мозга;
Биопсия скелетных мышц: выявляет диффузную гибель мышечных волокон и их замещение жировыми и фиброзными клетками.
Лабораторно-генетическое исследование.
Лечение:
Эффективного лечения нет.
Ведение больного направлено на предотвращение развития контрактур, поддержание мышечной силы и продление периода самостоятельного передвижения больного. Рекомендуются регулярные занятия лечебной гимнастикой (но без значительных нагрузок), массаж и при необходимости ортопедические мероприятия.
Курсовой прием преднизолона (в дозе 0,75 мг/кг массы тела) может на время увеличить мышечную силу, но не замедляет прогрессирования заболевания. Используют коэнзим
Q10.
МИОТОНИЯ
Миотонии — наследственные заболевания, проявляющиеся замедленным расслаблением скелетных мышц после их сокращения (миотонические феномены).
МИОТОНИЯ ТОМСЕНА:
Аутосомно-доминантное заболевание;
Первый признак: изменение голоса при плаче ребенка, он начинает задыхаться, а после плача лицо очень медленно расслабляется.
Протекает мягко, не уменьшая продолжительности жизни и не приводя к инвалидности.
Мышечная сила нормальна или даже повышена.
МИОТОНИЯ БЕККЕРА:
Аутосомно-рецессивное заболевание;
Развивается позже (4-12 лет), чем миотония Томсена;
Заболевание начинается с ног и постепенно генерализуется.
183

Мышечная слабость обнаруживается в начале движений после отдыха и исчезает при их повторении, примерно у 60% больных определяется постоянная слабость в мышцах предплечий, грудино-ключично-сосцевидных мышцах.
Протекает тяжело.
МИОТОНИЧЕСКАЯ ДИСТРОФИЯ:
Аутосомно-доминантное мультисистемное заболевание;
Характеризуется миотонией, мышечной атрофией, катарактой, сердечной аритмией.
Наиболее часто симптомы возникают в возрасте 15—35 лет.
Типичны двусторонний птоз, слабость и атрофии в дистальных отделах конечностей, мимических и жевательных мышцах, угасание сухожильных рефлексов.
Больные жалуются на напряжения мышц, затруднения при движениях кисти и при ходьбе, нарастающие на холоде.
Катаракта обычно развивается в 25—50 лет у 85% больных.
Нередко обнаруживается кардиомиопатия с поперечной блокадой, приступами Морганьи—Адамса—Стокса и сердечной недостаточностью.
Заболевание медленно прогрессирует и нередко приводит к инвалидности через
15—20 лет.
Диагностика:
Анализ крови: повышение КФК;
Электромиографии — снижение длительности и амплитуды потенциалов двигательных единиц, отсутствие признаков поражения периферических нервов или мотонейронов передних рогов спинного мозга;
Лечение:
Для уменьшения степени миотонии можно использовать дифенин, нифедипин или
диакарб.
ПЕРИОДИЧЕСКИЙ ПАРАЛИЧ (ПАРОКСИЗМАЛЬНАЯ МИОПЛЕГИЯ)
Периодический паралич — наследственное (аутосомно-доминантное) заболевание, проявляющееся приступами вялого (периферического) паралича скелетных мышц.
Патогенез: развитие параличей связывается с нарушением возбудимости мышечных мембран вследствие патологии кальциевых (гипокалиемическая форма) или натриевых каналов (гиперкалиемическая форма).
Клиническая картина:
Заболевание дебютирует в детском или юношеское возрасте.
184

Периодически в виде приступов возникают парезы конечностей мышц туловища и шеи с выраженной их гипотонией, угасанием сухожильнх рефлексов.
Парезы сохраняются от часа до нескольких дней, затем мышечная сила постепенно восстанавливается.
Гипокалиемические формы нередко возникают в состоянии покоя (утром или ночью) после предшествующей физической нагрузки или употребления богатой углеводами пиши.
Гиперкалиемические приступы протекают в более легкой форме и менее длительно, они могут быть вызваны охлаждением, сопровождаются парестезиями в области лица и конечностей.
Диагностика:
ОАК: снижение уровня калия (гипокалиемическая) или его повышение (гиперкалиемическая форма);
Дифференциальный диагноз необходимо провести с: болезнью Конна, тиреотоксикозом, болезнью Аддисона, заболеваниями желудочно-кишечного тракта, сопровождающиеся диареей, рвотой и мальабсорбцией, последствиями приема тиазидных диуретиков кортикостероидов или слабительных средств).
Лечение:
Гипокалиемическая форма:
Во время приступа назначают хлорид калия, при отсутствии эффекта еще 5 г через 1—2 часа.
С целью профилактики приступов рекомендуется диета с низким содержанием углеводов и натрия, но с высоким содержанием калия (курага, чернослив, картофель и др.)
При часто повторяющихся приступах используется диакарб.
Гиперкалиемическая форма:
При необходимости в/в вводится 20 мл 10% глюконата кальция;
С целью профилактики приступов рекомендуется диета с низким содержанием углеводов и калия, при частых приступах назначают тиазидные диуретики.
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Боковой амиотрофический склероз — быстро прогрессирующее нейродегенеративное заболевание нервной системы, обусловленное избирательным поражением мотонейронов спинного мозга, двигательных ядер бульбарной группы черепных нервов, корковых нейронов, кортикоспинальных трактов в головном мозге и боковых канатиках спинного мозга.
Этиология: заболевание связано с мутациями в гене супероксиддисмутазы-1;
185

Патогенез: гибель центральных и периферических нейронов обусловлена механизмами глутаматной эксайтоксичности, оксидативного стресса, дефектом нейротрофической функции и другими процессами, приводящими к апоптозу клеток.
Клиническая картина:
В зависимости от локализации клинически проявляющегося патологического процесса могут развиваться бульбарные нарушения с дисфагией, дизартрией, слабостью, атрофией и фасцикуляциями в мышцах языка, к которым скоро присоединяются нарушения дыхания.
Бульбарная форма (самая неблагоприятная):
Чаще возникают парезы кистей, стоп с атрофией мышц и фасцикуляциями в них, сопровождающиеся крампи (синдром внезапных непроизвольных болезненных сокращений отдельных мышц или мышечных групп продолжительностью от нескольких секунд до нескольких минут), оживлением сухожильных рефлексов, пирамидными стопными знаками, рефлексами орального автоматизма (шейногрудная или пояснично-крестцовая формы).
Со временем патологический процесс генерализуется, нарастает атрофия мышц, угасают рефлексы, развиваются нарушения глотания и дыхания, чаще всего приводящие к смерти.
Нередки нервно-психические нарушения в виде нарушения внимания, мышления, эмоциональных расстройств (депрессия).
Заболевание развивается постепенно, проявляясь слабостью мышц конечностей, мышечными подергиваниями (фасцикуляциями) и локальными атрофиями мышц или нарушением глотания (дисфагия) и речи (дизартрия), дыхания.
Большинство (90%) больных умирают в среднем через 3—5 лет после начала заболевания, в основном от дыхательных нарушений и присоединяющейся, часто аспирационной, пневмонии.
Диагностика:
Электромиографиея: выявляются уменьшение числа двигательных единиц, фасцикуляции, денервационная активность (потенциалы фибрилляций, положительные острые волны), значительное увеличение амплитуды и длительности двигательных единиц.
КТ или МРТ головного мозга: проводят для исключения других возможных заболеваний нервной системы.
При микроскопическом исследовании выявляется гибель пирамидных клеток прецентральной извилины, коры лобной доли больших полушарий головного мозга, нейронов двигательных ядер бульбарной группы черепных нервов, передних рогов
186

спинного мозга. Ядра глазодвигательных нервов и нейроны передних рогов крестцовых сегментов не поражаются, что обусловливает сохранность глазодвигательных и тазовых функций. Повреждаются аксоны и миелин кортикоспинальных трактов на уровне головного и спинного мозга, истончаются передние корешки спинного мозга, происходит атрофия мышц.
Лечение:
Эффективного лечения нет.
Применение ингибитора высвобождения глутамата рилузола (рилутека) по 100 мг в сутки несколько задерживает прогрессирование болезни и продлевает жизнь больных на несколько месяцев.
В качестве симптоматической терапии при спастичности можно использовать миорелаксанты (баклофена, сирдалуда),
При болезненных мышечных спазмах ног (крампи) —дифенин или реланиум,
При нередко возникающей депрессии и насильственном плаче —амитриптилин на ночь,
При нарушении глотания —калимин и установка назогастрального зонда.
При выраженной дыхательной недостаточности может быть использована искусственная вентиляция легких, которая существенно продлевает продолжительность жизни, однако при этом качество жизни больного остается крайне низким.
ПОЛИМИОЗИТ, ДЕРМАТОМИОЗИТ
Полимиозит — наиболее частая форма приобретенных воспалительных миопатий. При дерматомиозите помимо мышц поражается кожа.
Этиология и патогенез:
Предполагается дизиммунный характер заболевания с образованием антител, повреждающих мелкие сосуды кожи и мышц при дерматомиозите и мышечные волокна (образование Т-клеток) при полимиозите.
Полимиозит и дерматомиозит часто (от трети до половины случаев) развиваются на фоне других системных заболеваний соединительной ткани (системной красной волчанки, узелкового периартериита, ревматоидного артрита, склеродермии).
Клиническая картина:
Полимиозит:
Характерно постепенное, в течение недель и месяцев, нарастание слабости в проксимальных отделах конечностей, мышцах плечевого и тазового пояса.
Слабость ног проявляется затруднением ходьбы по лестнице, вставания из глубокого кресла, с колен или корточек.
187

При поражении мышц плечевого пояса и проксимальных отделов рук больному сложно поднять руки выше головы: причесать волосы или положить какой-либо предмет на верхнюю полку.
Из-за слабости задних мышц шеи больным бывает трудно удерживать голову, она свисает кпереди, возможно присоединение дисфагии и дисфонии.
Сухожильные рефлексы в паретичных конечностях обычно снижены, атрофия мышц не выражена.
Иногда отмечаются спонтанные боли в мышцах и их болезненность при пальпации.
Дерматомиозит:
Дерматомиозит имеет более острое течение по сравнению с полимиозитом.
Наряду с поражением мышц возникают яркие проявления патологии кожи в виде сыпи на теле, лице, конечностях, уплотнение кожи на пальцах рук.
Характерны лиловое окрашивание спинки носа, щек, лба и вокруг ногтей, а также незначительный отек вокруг глаз и рта.
Кожные проявления могут предшествовать поражению мышц, возникать одновременно с ними или позднее.
Диагностика:
Анализ крови: повышение КФК, СОЭ. В сыворотке крови часто наблюдается увеличение содержания миоглобина, глутамата и пирувата, активности лактатдегидрогеназы и альдолазы. Примерно у половины больных выявляют ревматоидный фактор и антинуклеарные антитела, у четверти больных — антитела к РНК-синтетазе.
Электромиография: обнаруживают характерные для миопатии изменения (снижение амплитуды и длительности потенциалов двигательных единиц), при этом часто регистрируют фибрилляции, что свидетельствует о поражении терминальных ветвей аксонов двигательных нейронов.
Биопсия мышц: обнаруживает воспалительный характер изменений.
Лечение:
Преднизолон;
При отсутствии эффекта от перорального приема преднизолона можно его сочетать с цитостатиками — азатиоприном или метотрексатом и др., а также использовать пульс-терапию метилпреднизолоном в течение 3—5 дней с последующим переходом на пероральный прием преднизолона и цитостатика.
Используют плазмаферез и внутривенное введение иммуноглобулина.
Для предотвращения стойких контрактур показана лечебная гимнастика.
Вбольшинстве случаев через 1—2 месяца лечения наблюдается нарастание мышечной силы, уменьшение степени пареза.
188
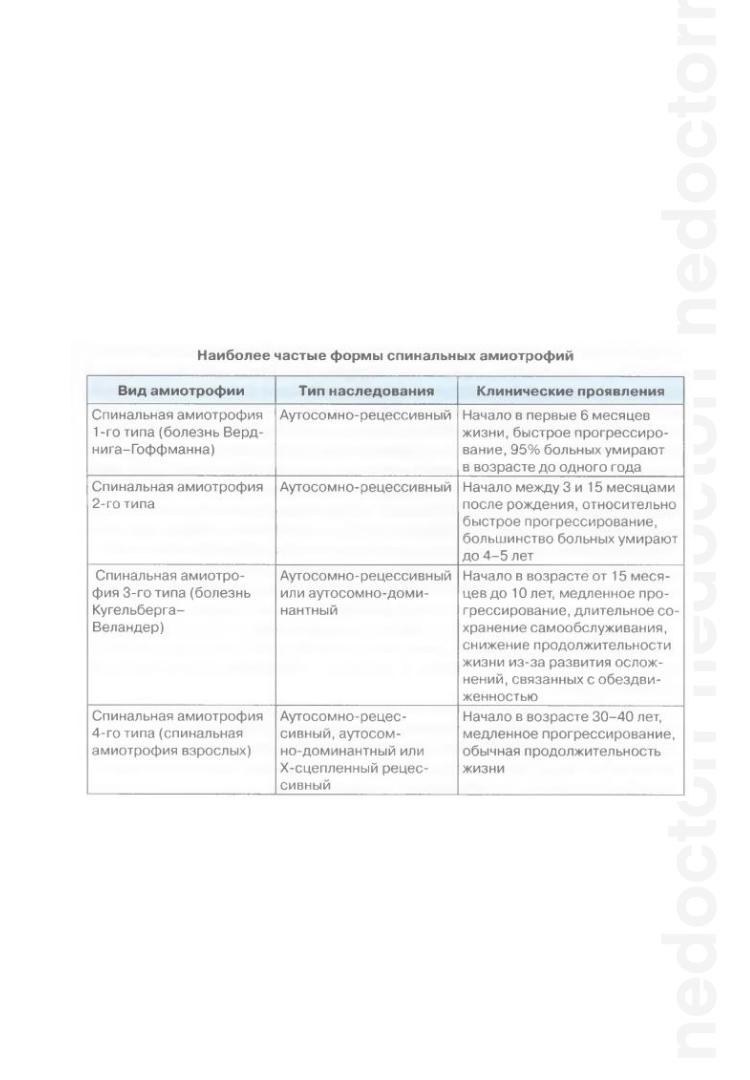
СПИНАЛЬНЫЕ АМИОТРОФИИ
Спинальные амиотрофии — наследственные заболевания, проявляющиеся прогрессирующими вялыми парезами и атрофией мышц вследствие поражения нейронов передних рогов спинного мозга. В мышцах могут наблюдаться фасцикуляции, чувствительных расстройств не отмечается.
Различные виды спинальных амиотрофий отличаются главным образом локализацией мышечной слабости, возрастом начала, скоростью прогрессирования заболевания, характером генетического дефекта и типом его наследования.
Диагноз подтверждается электромиографией, биопсией мышц и лабораторными генетическими исследованиями.
Эффективного лечения нет, показана лечебная гимнастика.
189

23. Рассеянный склероз.
Рассеянный склероз – хроническое аутоиммунное заболевание, характеризующееся повреждением и демиелинизацией ЦНС (головного и спинного мозга) с последующим аксональным повреждением.
Чаще страдают: люди молодого и среднего возраста (в 16-50 лет, но чаще в 20-25). Но чаще всего страдают женщины 20-30 лет, а также у людей с дефицитом витамина D.
Этиология:
Рассеянный склероз является мультифакториальным заболеванием, обусловленным различными внешнесредовыми воздействиями:
Вирусные или другие инфекции;
Экологические факторы;
Географические факторы;
Эндокринные факторы;
Наследственная предрасположенность.
Патогенез:
Мишень болезни: аксоны центральных проводящих путей и серое вещество больших полушарий головного мозга. при этом протекают 2 взаимосвязанных процесса:
1.Многоочаговое воспаление, затрагивающее миелин. При воспалении (аутоиммунном) развиваются очаги демиелинизации. Повреждение миелина полностью или частично нарушает проведение импульса по проводящим путям. Однако очаги демиелинизации могут подвергнуться обратному развитию (ремиелинизации) с устранением симптомов либо привести к формированию глиозного рубца.
2.Нейродегенерация – необратимое поражение центральных аксонов и гибель нейронов. Нейродегенерация развивается вследствие многоочагового воспаления и затрагивает аксоны центральных проводников и серое вещество головного и спинного мозга, вызывая стойкую и многоочаговую полиморфную симтоматику.
Очаги разрушения миелина обычно локализуются:
Вокруг желудочков ГМ;
В белом веществе спинного мозга;
В мозжечке;
В стволе ГМ;
В зрительных нервах.
На более поздних стадиях рассеянного склероза развивается атрофия головного мозга.
Клиническая картина:
По течению выделяют:
Ремиттирующим склероз (с периодами обострений и ремиссий). При волнообразном течении после обострения может наблюдаться полный или частичный регресс неврологических нарушений и затем следует ремиссия различной длительности (до нескольких лет).
190