

6 курс / Госпитальная педиатрия / Ответы
.pdfРазличают две формы заболевания:
-первичный идиопатический синдром Фанкони, в большинстве случаев носящий спорадический характер; единичные случаи могут являться наследственными (аутосомнорецессивное, аутосомно-доминантное наследование);
-вторичный синдром Фанкони, обусловленный генетическими болезнями (цистиноз, галактоземия, наследственная непереносимость фруктозы, тирозинемия (тип I), гликогеноз (тип I), болезнь Вильсона-Коновалова, митохондриальные цитопатии, болезнь Дента, синдром Лоу), токсическим действием лекарств (гентамицин, тетрациклин, антиретровирусные препараты), солей тяжелых металлов, либо развивающийся вследствие первичного амилоидоза, множественной миеломы и некоторых других заболеваний. Наиболее частой причиной синдрома Фанкони у детей является цистиноз, редкое аутосомно-рецессивное заболевание, которое характеризуется накоплением кристаллов цистина внутри лизосом и сопровождается прогрессирующим поражением интерстициальной ткани почек с исходом в хроническую почечную недостаточность.
Классификация 1. Болезнь де Тони–Дебре–Фанкони (первичная наследственная тубулопатия). 2. Синдром де Тони–Дебре–Фанкони, встречающийся при многих врожденных и приобретенных заболеваниях у детей Патогенез: Высказывается предположение, что в основе заболевания лежит либо
ферментативный дефект, либо аномалии транспортных белков почечных канальцев, а также генетически детерминированные аномалии проксимальных канальцев.
Клиника: Болезнь де Тони–Дебре–Фанкони характеризуется глюкозурией, фосфатурией, гипофосфатемией и аминоацидурией. Может встречаться в виде неполного синдрома, когда отсутствует один из этих симптомов. Основные клинические проявления (на первом году жизни, чаще во втором полугодии): жажда, полиурия, рвота, иногда запоры и длительный субфебрилитет. На втором году жизни отмечается резкое отставание в физическом развитии, костные деформации (вальгусные или варусные) нижних конечностей и других частей скелета, выраженная гипотония мышц, гипорефлексия, снижение АД. Полидипсия, повышения температуры тела без видимых причин, возможно, результат дегидратации. Потери калия с мочой приводят к развитию гипокалиемии с соответствующей клиникой (мышечная гипотония, гипорефлексия, снижение АД, изменения на ЭКГ). С метаболическим ацидозом, обусловленным нарушением реабсорбции бикарбонатов и калия, связаны признаки интоксикации: вялость, раздражительность, бледность кожных покровов. Рахитоподобная остеопатия — следствие сочетанного действия метаболического ацидоза, гипофосфатемии и дефицита кальция. Резистентность к витамину Д, по-видимому, обусловлена нарушением превращения его в активные формы в условиях метаболического ацидоза.
Лечение: Основными в лечении болезни ТДФ являются: 1) диетотерапия; 2) коррекция электролитов и КОС; 19 3) назначение витамина Д и его метаболитов.
1. Диета предусматривает: – ограничение потребления продуктов, содержащих ацидогенные аминокислоты (яйца, творог); – включение в рацион продуктов, богатых фосфатами, калием (чернослив, курага, изюм); – потребление продуктов, оказывающих ощелачивающее действие (молоко, молочные продукты, фруктовые соки); – неограниченное потребление жидкости. Лучше всего придерживаться картофельной или капустной диеты.
2. Для коррекции электролитов и КОС показано: – при выраженном ацидозе — введение щелочных растворов (бикарбоната натрия, цитратных смесей) (см. раздел ПКА); – при дефиците калия — панангин или аспаркам в возрастных дозировках; при выраженной гипокалиемии противопоказано парентеральное введение глюкозы (раствора более 10 %), особенно в условиях ацидоза.
3. Для ликвидации нарушений фосфорно-кальциевого обмена применяется витамин Д и его метаболиты при постоянном контроле уровня кальция в моче и крови. Начальная доза витамина Д — 10 000–15 000 МЕ/сут. Повышение дозы — каждые две недели на 10 000–15 000 МЕ до нормализации показателей Са и Р в крови и моче. Максимальная доза — 75 000–100 000 МЕ/сут. Из метаболитов витамина Д рекомендуются: оксидевит в дозе 0,5– 1,5 мкг/сут, кальцитриол, альфакальцидол — 0,5–1 мкг/сут, препараты кальция (глюконат кальция — 1,5– 2,0 мкг/сут), препараты фосфора (фитин — 0,5–1,0 мкг/сут), остеогенон — 1 таб. 3 раза в день (прил. 6). Лечение проводится при постоянном контроле (каждые 10–14 дней) уровня кальция, фосфора, активности щелочной фосфатазы, калия, КОС крови.
66. Тубулопатии. Почечный тубулярный ацидоз: причины, патогенез, клинические
варианты.
Почечный тубулярный ацидоз - многочисленная группа заболеваний, в основе которых лежит неспособность почечных канальцев подкислять мочу и сопровождающихся сдвигом реакции крови в кислую сторону. В конечном итоге патология характеризуется метаболическим ацидозом, полиурией, рахитоподобными изменениями, нефрокальцинозом и нефролитиазом.
Причины: цистиноз, галактоземия, гликогеноз (тип I), тирозинемия, болезнь Вильсона, гиперпаратиреоидизм, медуллярная кистозная болезнь, витамин-Д-дефицитный и зависимый рахит, идиопатическая гиперкальциурия, первичная гипероксалурия, поражение проксимальных канальцев солями тяжелых металлов, некоторыми лекарственными препаратами.
Патогенез: в норме в проксимальных канальцах реабсорбируется до 90% профильтрованных бикарбонатов. Вследствие нарушения реабсорбции бикарбонатов в проксимальном канальце, бикарбонатурия развивается при нормальной концентрации бикарбонатов в плазме крови. Это ведёт к метаболическому ацидозу при отсутствии подкисления мочи, несмотря на сохранные механизмы дистальной секреции ионов H + . Как только концентрация плазменных бикарбонатов снижается ниже порогового значения (в большинстве случаев менее 15 ммоль/л, в отсутствии лечения), профильтрованные бикарбонаты начинают полностью реабсорбироваться, реакция мочи становится кислой.
Почечный тубулярный ацидоз I типа (дистальный, классический). При этом варианте дистальный каналец не способен создавать определенный градиент концентрации водородных ионов между кровью и канальцевой жидкостью, нарушена их секреция в дистальных канальцах и снижено содержание. Особенностью этой формы является то, что в мочу постоянно выделяются гидрокарбонаты, вместо которых в кровь диффундируют ионы хлора — развивается гиперхлоремия.
Для классического ПКА I типа характерно: – отставание в росте; – рахитоподобные изменения в костях; – метаболический ацидоз; – кризы обезвоживания и полиурия; – нефрокальциноз (двусторонний) и мочекаменная болезнь с тубулоинтерстициальным нефритом и/или пиелонефритом; – повышение уровня хлоридов крови; – гипокалиемия; – щелочная реакция мочи (рН = 6,7–7,2); – гипоцитратурия; – гиперкальциурия до 10–20 мг/сут (при норме 1–5 мг/сут); Гипокальциемия стимулирует выработку ПТГ, развивается вторичный гиперпаратиреоз, что приводит к резорбции костной ткани. В результате этого в кровь поступает кальций, поэтому отмечается нормокальциемия. Развиваются деформации (Х-образные искривления нижних конечностей, бочкообразная форма грудной клетки и др.).
Почечный тубулярный ацидоз II типа (проксимальный канальцевый ацидоз). В его основе лежит повышенная экскреция бикарбонатов с мочой и нарушение их реабсорбции в проксимальных канальцах. Клинически это заболевание проявляется в 3–18 мес. с таких симптомов, как: – полидипсия, полиурия; – повышение температуры неясного генеза; – отставание в физическом развитии; – деформации большеберцовых и бедренных костей; – метаболический ацидоз (снижение бикарбонатов и рН); – гиперхлоремия; – гипокалиемия; – резкое снижение порога реабсорбции бикарбонатов; – гипоили нормокальциемия; 13 – щелочная реакция мочи за счет повышенной экскреции бикарбонатов с мочой, но может быть кислая и нейтральная.
Лечение: коррекция метаболического ацидоза осуществляется введением растворов натрия бикарбоната. Надо помнить, что быстрая коррекция может быть опасной из-за компенсаторного дыхательного алкалоза, поэтому устранять метаболический ацидоз нужно поэтапно: в первые 12 часов — лишь частично (примерно на 1 /3), оставшийся дефицит — в течение следующих 6 часов.
Для уменьшения ацидоза показана цитратная смесь (140 г лимонной кислоты, 90 г кристаллической соли цитрата натрия на 1 л воды) — по 15–25 мл 3 раза в день. Препараты калия: при I типе ПКА — 2 ммоль/кг/сут, при II типе — 4–10 ммоль/кг/сут при контроле уровня К в сыворотке крови (нормальный уровень К — 3,5–5,1 ммоль/л). При гипокальциемии — препараты кальция. При остеопорозе и остеомаляции (деформации костной системы): – витамин Д — начальные суточные дозы 10 000–20 000 МЕ, максимальные — 30 000–60 000 МЕ; – метаболиты витамина Д (оксидевит) — по 0,5–2 мкг/сут.
68. Первичные иммунодефицитные состояния у детей: синдром Вискотта-Олдрича. Причины, клиника, диагностика, лечение.
Синдром Вискотта-Олдрича (СВО) - это Х-сцепленное заболевание, характеризующееся комбинированным иммунодефицитом в сочетании с тромбоцитопенией и экземой. Заболевание является результатом мутации гена, кодирующего белок WASP, который принимает участие в полимеризации актина и формировании цитоскелета. Отсутствие белка WASP в лимфоцитах и тромбоцитах больных приводит к развитию тромбоцитопении, нарушению функций Т-клеток и регуляции синтеза антител.
Клиника: начинается в младенческом или раннем детском возрасте; -геморрагический синдром, нередко очень тяжелый – петехиальная сыпь, кефалогематома, кровотечения из пупочной ранки, кишечные кровотечения.
-экзема – у 80% больных;
-повторные, как правило, необычные (тяжелые герпетические инфекции, пневмоцистные пневмонии) и плохо поддающиеся коррекции вирусные, бактериальные и грибковые инфекции ЛОРорганов, дыхательной системы, органов пищеварения, кожи.
-у 70% больных выявляют аутоиммунные заболевания (гемолитическая анемия, нейтропения, артрит, кожный васкулит, неспецифический язвенный колит, церебральный васкулит, гломерулонефрит, аутоиммунная тромбоцитопения)
-у больных старше 5 лет повышена частота злокачественных новообразований (преимущественно опухоли лимфоидной ткани)
Диагностика: -тромбоцитопения, уменьшение размеров тромбоцитов, экзема, мужской пол, наличие семейного анамнеза, комбинированный ИДС.
Иммунологические изменения: лимфопения, в основном за счет Т-лимфоцитов, снижение функциональной активности Т-клеток, первоначально нормальный уровень сывороточных иммуноглобулинов затем прогрессивно снижается (в первую очередь за счет иммуноглобулинов М); нарушена продукция антител, особенно к полисахаридным антигенам. Лечение: единственным методом излечения больных является трансплантация костного мозга (ТКМ) от HLA-идентичного донора.
При отсутствии возможности ТКМ показано проведение спленэктомии, так как это приводит к значительному уменьшению геморрагического синдрома.
После спленэктомии необхома постоянная терапия противопневмококковыми антибиотиками (антибиотики пенициллинового ряда, например, бициллин)
-заместительная терапия внутривенным иммуноглобулином
-постоянная профилактическая антибактериальная (триметоприм – сульфаметоксазол) противовирусная (ацикловир в поддерживающей дозе) и противогрибковая (флуконазол) терапия -для лечения аутоиммунных расстройств – глюкокортикоиды, циклоспорин А
-необходима симптоматическая терапия экземы и других аллергических заболеваний
-переливание тромбоцитов только для купирования тяжелых кровотечений
-возможна вакцинация инактивированными вакцинами или анатоксинами.
68. Первичные иммунодефицитные состояния у детей: синдром Вискотта-Олдрича.
Причины, клиника, диагностика, лечение.
Синдром Вискотта-Олдрича (СВО) - Х сцепленный комбинированный первичный иммунодефицит, для которого, помимо иммунологических нарушений, характерны тромбоцитопения с малым размером тромбоцитов и атопический дерматит. Как и для других комбинированных иммуноефицитов, для СВО также характерна высокая частота аутоиммунных и опухолевых осложнений. Синдромом Вискотта -Олдрича страдают лица мужского пола.
Причины: мутация гена WASP. Белок WASP экспрессируется исключительно в клетках гематопоэтического ряда. WASP необходим для реорганизации клеточного цитоскелета, что играет важную роль в таких функциях клетки, как формирование иммунологических синапсов, движение клеток, а также перемещение белков внутри клетки. Мутации гена приводят к значительному снижению или полному отсутствию белка WASP. Степень выраженности проявлений варьирует между больными, и частично коррелирует с концентрацией белка в клетках, что в свою очередь, зависит от локализации и вида мутации. Имеющиеся исследования
позволяют предположить, что тромбоцитопения при СВО в основном связана с повышенным разрушением тромбоцитов.
Клиника:
Общая характеристика:
Тяжесть проявлений заболевания у больных с СВО варьирует от интермиттирующей тромбоцитопении с минимальными геморрагическими проявлениями до тяжелого заболевания с выраженным инфекционным и аутоиммунным синдромами.
Геморрагический синдром Поскольку тромбоцитопения, как правило, отмечается с рождения, заболевание может
манифестировать кровотечениями из пупочной ранки, а также такими симптомами, как мелена, эпистаксис, гематурия, петехиальная сыпь, а также жизнеугрожающими внутричерепными и желудочно-кишечными кровотечениями. Кровотечения являются основной причиной летальности при СВО. Атопический дерматит различной степени выраженности проявляется, как правило, на первом году жизни и часто сопровождается местным инфицированием. У больных с легким течением СВО дерматит может отсутствовать или носить легкий, преходящий характер.
Инфекционные проявления У большинства больных СВО с возрастом проявляются прогрессирующие признаки
иммунодефицита. Вследствие нарушений гуморального и клеточного иммунитета, у больных со среднетяжелым или тяжелым течением СВО наблюдаются частые инфекции, которые нередко встречаются в первые шесть месяцев жизни. Из них наиболее распространенны отиты, синуситы и пневмония.
Аутоиммунные нарушения Аутоиммунные нарушения характерны для тяжелого течения болезни. Некоторые больные
развивают более одного аутоиммунного заболевания.
Нередко больные СВО развивают иммунную тромбоцитопению. Злокачественные новообразования
Злокачественные новообразования чаще развиваются у взрослых или подростков с СВО, но могут встречаться и у детей (чаще всего лимфомы).
Диагностика:
Анамнез При сборе семейного анамнеза надо обращать внимание на случаи повторных тяжелых
инфекций и\или смертей мальчиков в раннем возрасте с клиникой инфекционных заболеваний и\или кровотечений. При опросе родителей следует уточнить сроки возникновения, частоту и тяжесть проявления геморрагического синдрома (кожная сыпь, кровотечения, стул с кровью), инфекционных заболеваний у ребенка, наличия и выраженности атопического дерматита, сроки и частоту госпитализаций в стационары.
Физикальное обследование Пациенты с СВО могут отставать в росте. Важно обратить внимание на наличие петехий,
экхимозов, проявлений атопического дерматита, наличия инфицированных очагов. Для больных с СВО характерна локализованная или генерализованная лимфоаденопатия. У больных с СВО нередко отмечается гепатоспленомегалия.
Лабораторная диагностика:
Анемия неясного генеза, тромбоцитопения, Исследование внутриклеточной экспрессии белка WASP позволяет не только с большой долей вероятности подтвердить диагноз, и предположить прогноз заболевания в 8 зависимости от его концентрации. Больные с наличием экспрессии белка WASP имеют более благоприятный прогноз.
Учитывая комбинированный иммунодефицит, необходимо контролировать вирусные инфекции, в первую очередь, цитомегаловирусную виремию, для чего рекомендовано использовать метод количественой полимеразной цепной реакции ( ПЦР). Копрология и кал на скрытую кровь при подозрении на гемоколит. Проведение молекулярно-генетического анализа
Анализ на выявление мутации гена WASP проводится с помощью полимеразной цепной реакции и последующего секвенирования продуктов.
Инструментальная диагностика Ультразвуковое исследование брюшной полости и забрюшинного пространства для выявления
спленомегалии. Компьютерная томография органов брюшной и грудной полости - по показаниям. При наличии острой инфекционной симптоматики - рентгенография грудной клетки, придаточных пазух носа в динамике. Другие инструментальные исследования - при наличии соответствующих клинических показаний. Дифференциальный диагноз должен проводиться с иммунной тромбоцитопенией, которая может являться как самостоятельным заболеванием, так и сопровождать течение других иммунодефицитных состояний (например, гипер-IgM синдром).
Лечение:
Метод выбора: трансплантация гемопоэтических стволовых клеток.
Спленэктомия снижает вероятность кровотечений, но сопровождается повышенным риском септицемии и в настоящее время не рекомендуется. Хорошо себя зарекомендовала терапия агонистом тромбопоэтиновых рецепторов ромипластином, что является хорошей альтернативой коррекции тромбоцитопении. Препарат используется в дозе 9-10 мг\кг 1 раз в неделю. Переливания тромбоцитов следует избегать, если у больного нет серьезной угрозы для жизни, и кровотечение может быть остановлено консервативными способами. Кровоизлияния в ЦНС требуют немедленного переливания тромбоцитов. Тромбоциты и другие продукты крови должны подвергаться облучению перед переливанием для предотвращения реакции трансплантат против хозяина. Так как у больных СВО наблюдается нарушение продукции антител в ответ на многие разновидности антигенов, то профилактическое лечение с помощью внутривенного переливания иммуноглобулинов (ВВИГ) показано практически всем больным СВО.
69. Синдром мальабсорбции. Целиакия: этиология, патогенез, клиника, прогноз.
Целиакия - хроническое генетически детерминированное заболевание, характеризующееся стойкой неперносимостью глютена с развитием гиперрегенераторной обратимой атрофии слизистой оболочки тонкой кишки и связанного с ней синдрома мальабсорбции. Этиология и патогенез: Этиопатогенез окончательно не изучен.
Предполагается аутосомно-доминантный характер передачи с неполной пенетрантностью. Значимость генетических факторов подтверждают изменения, выявляемые при дуоденальной биопсии у 10-15% близких родственников. Более 97% больных целиакией имеют маркеры гистосовместимости HLA-DQ2 и/или DQ8.
Глютен - группа белков, которые содержатся в пшенице, ячмене, овсе и ржи в виде клейковины. Наиболее токсичным компонентом является альфа-глиадин. Аномальная чувствительность к глютену вызывает повреждение слизистой оболочки тонкой кишки и воспалительную реакцию, которая при длительном течении вызывает постепенную атрофию ворсинок.
Глютен связывается со спецефическими рецепторами эпителиоцитов, детерминированными генами HLA и (или) поврежденными вирусами и активирует Th1-иммунный ответ слизистой оболочки. Увеличивается количество лимфоцитов и плазмоцитов, вырабатывающих антиглиадиновые антитела, а также глиадинспецифичных Т-лимфоцитов в собственной пластинке слизистой оболочки.
При прогрессировании процесса запускается аутоимунный механизм, в результате которого вырабатываются антиэндомизийные и антиретикулиновые антитела.
Морфологически выявляются:
-инфильтрация плазмоцитами и лимфоцитами собственного слоя слизистой оболочки;
-гиперплазия и углубление кишечных крипт;
-преимущественная атрофия (укорочение ворсинок) тонкой кишки.
Клинические проявления Гастроинтестинальные расстройства включают:
-диарею;
-рвоту;
-абдоминальную боль;
-метеоризм;
-анорексию;
-запор.
Диарея при целиакии характеризуется обильным пенистым зловонным стулом, стеатореей. Вздутие живота в сочетании с тонкими конечностями вследствие атрофии мышц создают "вид паука".
Синдром мальабсорбции служит причиной развития железодефицитной анемии (часто - единственный симптом), периферических отеков (в результате гипопротеинемии), потери массы тела при хорошем аппетите, болей в костях, парестезии, слабости, повышенной утомляемости.
Внекишечные проявления часто выражены в сочетании с минимальными интестинальными симптомами или без них:
-герпетиформный дерматит с интенсивной сыпью на поверхностях разгибательных мышц конечностей;
-необъяснимо низкий рост у детей, их отставание в физическом и половом развитии, снижение массы тела у взрослых (наличие ожирения не исключает диагноза целиакии);
-бесплодие или повторные выкидыши у женщин;
-развитие остеопороза, гиповитаминоза;
-белково-энергетическая недостаточность;
-рецидивирующий афтозный стоматит.
Могут наблюдаться гипоплазия зубной эмали, мигренозные головные боли, в крови повышено содержание трансаминаз. Отмечена ассоциация целиакии с аутоимунными эндокринопатиями, например тиреоидитом.
Атипичные (бессимптомные) формы целиакии встречаются у 1/3 больных с преобладанием внекишечной симптоматики:
-снижение массы тела;
-железодефицитная анемия;
-гипополивитаминоз;
-остеопороз;
-мышечные, костные, суставные боли, мышечная слабость;
-другие признаки гипокалиемии и гипокальциемии;
-вздутие живота, дискомфорт, метеоризм.
Прогноз благоприятен при своевременной диагностике и строгом соблюдении диеты. Улучшение наступает в течение нескольких дней после исключения из пищи глютена. Все симптомы обычно исчезают через 4-6 недель.
Смертность больных с клинически диагностированной целиакией вдвое выше, чем в контрольной популяции. Основная причина смерти - развитие лимфоретикулярной патологии (особенно лимфомы кишечника).
Четкой корреляции между частотой возникновения злокачественных опухолей желудочнокишечного тракта и соблюдением диеты не найдено.
70. Анафилактический шок: клинические формы, неотложная помощь.
Анафилактический шок (АШ) – острая системная аллергическая реакция на повторный контакт с аллергеном, угрожающая жизни и сопровождающаяся выраженными гемодинамическими нарушениями, а также нарушениями функций других органов и систем. СТАДИИ ФОРМИРОВАНИЯ: ИММУНОЛОГИЧЕСКАЯ, ПАТОХИМИЧЕСКАЯ, ПАТОФИЗИОЛОГИЧЕСКАЯ.
Клинические формы:
Молниеносная (10 минут), немедленная (дошоковый период 30-40 минут), замедленная (шок проявляется через несколько часов);
По клиническим вариантам
•типичный;
•гемодинамический (коллаптоидный);
•асфиксический;
•церебральный;
•абдоминальный.
По течению
•острое доброкачественное;
•острое злокачественное;
•затяжное;
•рецидивирующее;
•абортивное.
По степени тяжести
•I степень;
•II степень;
•III степень;
•IV степень. Неотложная помощь:
1.Прекратить действие аллергена на организм.
2.Уложить пациента на спину и приподнять ножной конец, голову повернуть набок.
3.Обеспечить доступ к вене, параллельно измерить АД, пульс.
4.При низком АД ввести адреналин 0,1% 1 мл в/в капельно.
5.В\в или в/м ввести кортикостероиды (преднизолон, дексаметазон).
6.Ввести п/к или в/м антигистаминный препарат (пипольфен 2-4 мл 2,5%, супрастин 2-4 мл 2%, димедрол 5 мл 1%).
71. Аллергический ринит у детей. Этиология. Патогенез. Клиническая картина.
Диагностика. Принципы терапии.
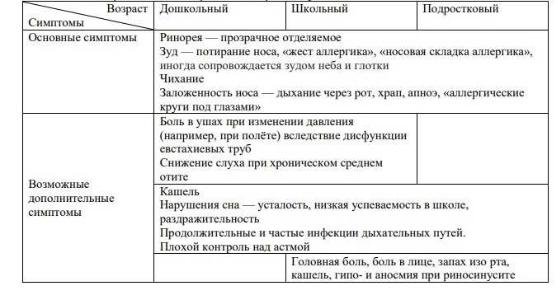
АР – IgE обусловленное воспалительное заболевание слизистой оболочки носа, проявляющееся комплексом симптомов в виде чихания, зуда, ринореи и заложенности носа.
Этиология:
Для классификации аллергенов используют несколько подходов:
• по пути поступления в организм (ингаляционные, энтеральные, контактные, парентеральные, трансплацентарные);
•по распределению в окружающей среде (аэроаллергены, аллергены помещений, аллергены внешние, промышленные и профессиональные аллергены и сенситизаторы);
•по категориям (инфекционные, тканевые, неинфекционные, лекарственные, химические);
•по происхождению (лекарственные, пищевые, аллергены насекомых или инсектные);
•по диагностическим группам (бытовые, эпидермальные, споры плесневых грибов, пыльцевые, инсектные, лекарственные и пищевые).
Для обозначения аллергенов разработана специальная международная номенклатура:
•неинфекционные — бытовые (аэроаллергены жилищ), эпидермальные, пыльцевые, пищевые, инсектные, лекарственные аллергены;
•инфекционные — грибковые, бактериальные аллергены.
Впатогенезе аллергических болезней реакции немедленного типа (IgE-зависимого, анафилактического, атопического) являются основными (но не всегда единственными). При первом контакте с аллергеном образуются специфические белки — IgE антитела, которые фиксируются на поверхности тучных клеток в различных органах. Это состояние называется сенсибилизация — повышение чувствительности к конкретному АлГ. При повторном контакте сенсибилизированного организма с причинным АлГ происходит развитие IgE-зависимого воспаления в слизистой оболочке носовой полости, обусловливающего появление симптомов. В большинстве случаев у одного пациента имеет место сенсибилизация одновременно к нескольким аллергенам, относящимся к разным группам.
Втечение первых минут после воздействия АлГ (ранняя фаза аллергической реакции) происходят активация тучных клеток и базофилов, дегрануляция и выделение медиаторов воспаления (гистамина, триптазы, простагландина D2, лейкотриенов, фактора активации тромбоцитов). В результате действия медиаторов повышается сосудистая проницаемость, увеличивается продукция слизи, сокращается гладкая мускулатура, возникают острые симптомы аллергических болезней: зуд глаз, кожи, носа, гиперемия, отек, чихание, водянистые выделения из носа.
Через 4–6 часов (поздняя фаза аллергической реакции) после воздействия АлГ происходит изменение кровотока, экспрессия молекул клеточной адгезии на эндотелии и лейкоцитах, инфильтрация тканей клетками аллергического воспаления — базофилами, эозинофилами, Т лимфоцитами, тучными клетками. В результате происходит формирование хронического аллергического воспаления, одним из клинических проявлений которого является неспецифическая тканевая гиперреактивность. Характерными симптомами являются назальная гиперреактивность и обструкция, гипо- и аносмия.
Клиническая картина:
Диагностика:
1.Мазок-отпечаток со слизистой носа. При наличии аллергии в анализе будет обнаружено повышение числа лимфоцитов, эозинофилов, тучных клеток.
2.Анализ на определение уровня IgE (антител к различным группам аллергенов) и уровня общего IgE.

3.Кожные пробы (аллерготесты). Во время исследования на кожу или под кожу наносятся группы аллергенов – возможных причин возникновения ринита. Наличие покраснения в области нанесения тех или иных аллергенов показывает истинную причину появления симптомов аллергии.
4.Риноскопию. При данном исследовании врач проверяет слизистые носа пациента на наличие в них проявлений сопутствующих заболеваний – полипов, гнойных выделений,
гипертрофии слизистой и др.
Лечение:
Основная цель терапии — достижение контроля над болезнью. Комплекс терапевтических мероприятий включает:
1.ограничение контакта с патогенетически значимыми аллергенами;
2.лекарственную терапию;
3.аллерген-специфическую иммунотерапию;
4.обучение.
Медикаментозное лечение: антигистаминные препараты, интраназальные глюкокортикостероиды, иммунотерапия.
72. Нарушение обмена белков. Фенилкетонурия. Этиопатогенез. Клинические проявления. Диагностика. Лечение.
Фенилкетонурия обусловлена дефицитом фермента фенилаланингидроксилазы, приводящим к накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии) и продуктов его распада. Заболевание вызвано мутацией гена фенилаланингидроксилазы (РАН), локализующегося на длинном плече хромосомы 12, участке 12q22-q24.1.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин- 4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Клиника
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда
– беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный

«мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко - врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика:
При отсутствии лечения на первом году жизни, обычно в возрасте 2−6 месяцев, родителей беспокоят вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги.
При отсутствии лечения обращают на себя внимание следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз, своеобразный «мышиный» запах мочи больных, возможно формирование микроцефалии. В психоневрологическом статусе отмечаются задержка статико-моторного и психоречевого развития, симптоматическая эпилепсия, а в некоторых случаях ― гидроцефалия. Эпилептические приступы встречаются почти у половины нелеченых больных и в некоторых случаях могут служить первым признаком болезни. Обычно отмечаются генерализованные пароксизмы по типу инфантильных спазмов в виде «салаамовых судорог», кивков; могут наблюдаться абсансы. Приступы носят упорный характер и плохо поддаются антиконвульсантной терапии. При отсутствии патогенетического лечения болезнь медленно прогрессирует.
Лабораторная диагностика: неонатальный скрининг (уровень ФА выше 2.0 мг/дл = ГФА, ГФА больше.10 мг/дл = ФКУ), проба Гатри (ФА в крови), проба Феллинга (моча на фенилпировиноградную к-ту, положительная с 6 недель),тест на потенциальную чувствительность к сапроптерина гидрохлориду.
Лечение:
Тактика терапии зависит от степени тяжести:
-Легка гиперфенилаланинемия требует наблюдения и диф.диагностики. Постоянное наблюдение врача, контроль уровня ФА.
-Умеренная (мягкая, средняя) ФКУ требует назначения гипоФА диеты, тест на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
-Классическая (тяжелая) форма требует строгой диеты и тест на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
ДИЕТА ПРИ УРОВНЕ ФА >/= 360 МКМОЛЬ/Л.