

54. Классификация, номенклатура и изомерия аминов. Алифатические и ароматические амины, первичные, вторичные и третичные амины.
Ответ.
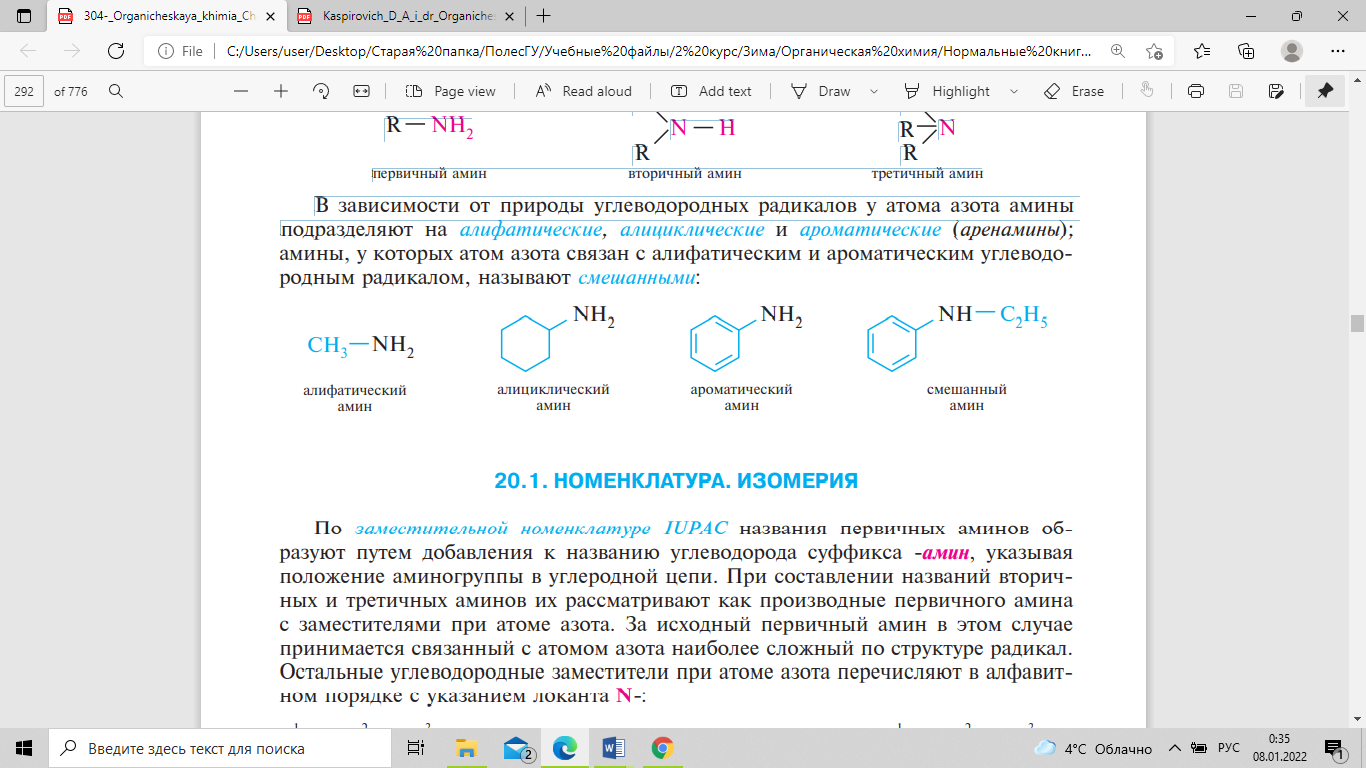
Аминами
называют производные аммиака, в молекуле
которого один, два или три атома водорода
замещены углеводородными радикалами.
Соответственно числу углеводородных
остатков различают первичные, вторичные
и третичные амины. В зависимости от
природы углеводородных радикалов у
атома азота амины подразделяют
на алифатические, алициклические и
ароматические (аренамины); амины, у
которых атом азота связан с алифатическим
и ароматическим углеводородным радикалом,
называют смешанными:
 По
заместительной номенклатуре
IUPAC названия первичных аминов образуют
путем добавления к названию углеводорода
суффикса -амин, указывая положение
аминогруппы в углеродной цепи. При
составлении названий вторичных и
третичных аминов их рассматривают как
производные первичного амина с
заместителями при атоме азота. За
исходный первичный амин в этом случае
принимается связанный с атомом азота
наиболее сложный по структуре радикал.
Остальные углеводородные заместители
при атоме азота перечисляют в алфавитном
порядке с указанием локанта N-:
По
заместительной номенклатуре
IUPAC названия первичных аминов образуют
путем добавления к названию углеводорода
суффикса -амин, указывая положение
аминогруппы в углеродной цепи. При
составлении названий вторичных и
третичных аминов их рассматривают как
производные первичного амина с
заместителями при атоме азота. За
исходный первичный амин в этом случае
принимается связанный с атомом азота
наиболее сложный по структуре радикал.
Остальные углеводородные заместители
при атоме азота перечисляют в алфавитном
порядке с указанием локанта N-:
 Если
соединение содержит две или три
аминогруппы, то в названии их обозначают
множительными приставками ди- или три-,
которые ставятся перед суффиксом –амин.
Простейшие амины чаще всего называют
по радикало-функциональной номенклатуре.
Согласно этой номенклатуре, названия
аминов образуют из названий углеводородных
радикалов, перечисляемых в алфавитном
порядке, и суффикса -амин:
Если
соединение содержит две или три
аминогруппы, то в названии их обозначают
множительными приставками ди- или три-,
которые ставятся перед суффиксом –амин.
Простейшие амины чаще всего называют
по радикало-функциональной номенклатуре.
Согласно этой номенклатуре, названия
аминов образуют из названий углеводородных
радикалов, перечисляемых в алфавитном
порядке, и суффикса -амин:
 Некоторые
амины сохраняют тривиальные названия:
Названия
первичных ароматических аминов, а также
смешанных аминов обычно образуют на
основе названия родоначального
представителя — анилина. В случае
смешанных аминов положение заместителей
у атома азота обозначают с помощью
локанта N-:
Некоторые
амины сохраняют тривиальные названия:
Названия
первичных ароматических аминов, а также
смешанных аминов обычно образуют на
основе названия родоначального
представителя — анилина. В случае
смешанных аминов положение заместителей
у атома азота обозначают с помощью
локанта N-:

Вторичные
и третичные ароматические амины, как
правило, называют по радикало-функциональной
номенклатуре. Изомерия
аминов обусловлена разной структурой
углеводородных радикалов, разным
положением аминогруппы и метамерией.
Сущность метамерии состоит в том, что
амины с одной и той же брутто-формулой
могут быть первичными, вторичными и
третичными. Приведенные соединения
являются метамерами:

55. Электронное строение аминов. Роль неподеленной электронной пары азота в проявлении основных и нуклеофильных свойств алкил- и ариламинов. Реакции ацилирования и алкилирования аминов. Аммониевые соли.
Ответ.
Алкиламинами
называют продукты замещения одного,
двух или трех атомов водорода в аммиаке
алкильными группами. Амины образуют
менее прочные ассоциаты, чем соответствующие
спирты, в связи с тем, что электроотрицательность
атома азота меньше, чем атома кислорода.
Третичные амины не имеют атома водорода
при атоме азота, поэтому они не способны
к ассоциации. Пространственная
модель молекулы алкиламина имеет форму
четырехгранной пирамиды, в вершине
которой находится атом азота. Валентные
углы между связями в среднем равны
107—108°, то есть близки к тетраэдрическому.
Условно приняв неподеленную пару
электронов атома азота за четвертый
заместитель, можно представить
конфигурацию атома азота в аминах
аналогично тетраэдрической конфигурации
атома углерода. Атом азота в аминах
находится в sp3-гибридизации, а неподеленная
пара электронов занимает sp3-гибридную
орбиталь. Такое строение предполагает
существование оптической изомерии у
соединений, имеющих три разных заместителя
у атома азота (роль четвертого заместителя
выполняет неподеленная пара электронов).
Реакционная способность алкиламинов
определяется главным образом наличием
у атома азота неподеленной пары
электронов. За счет пары электронов
атома азота амины, с одной стороны,
способны присоединять протон от кислоты,
проявляя при этом основные свойства, а
с другой — могут атаковать в молекуле
реагента электрофильный центр (чаще
атом углерода, несущий частичный или
полный положительный заряд) и образовывать
химическую связь с ним, проявляя
нуклеофильные свойства. С кислотами
алкиламины образуют соли алкиламмония:
![]() При
действии на аммониевую соль сильного
основания — натрия гидроксида
высвобождается исходный амин. Реакцию
солеобразования с последующим выделением
свободного амина часто используют для
очистки аминов. При взаимодействии с
галогеналканами первичные амины
превращаются во вторичные, вторичные
— в третичные, а третичные — образуют
четвертичные аммониевые соли:
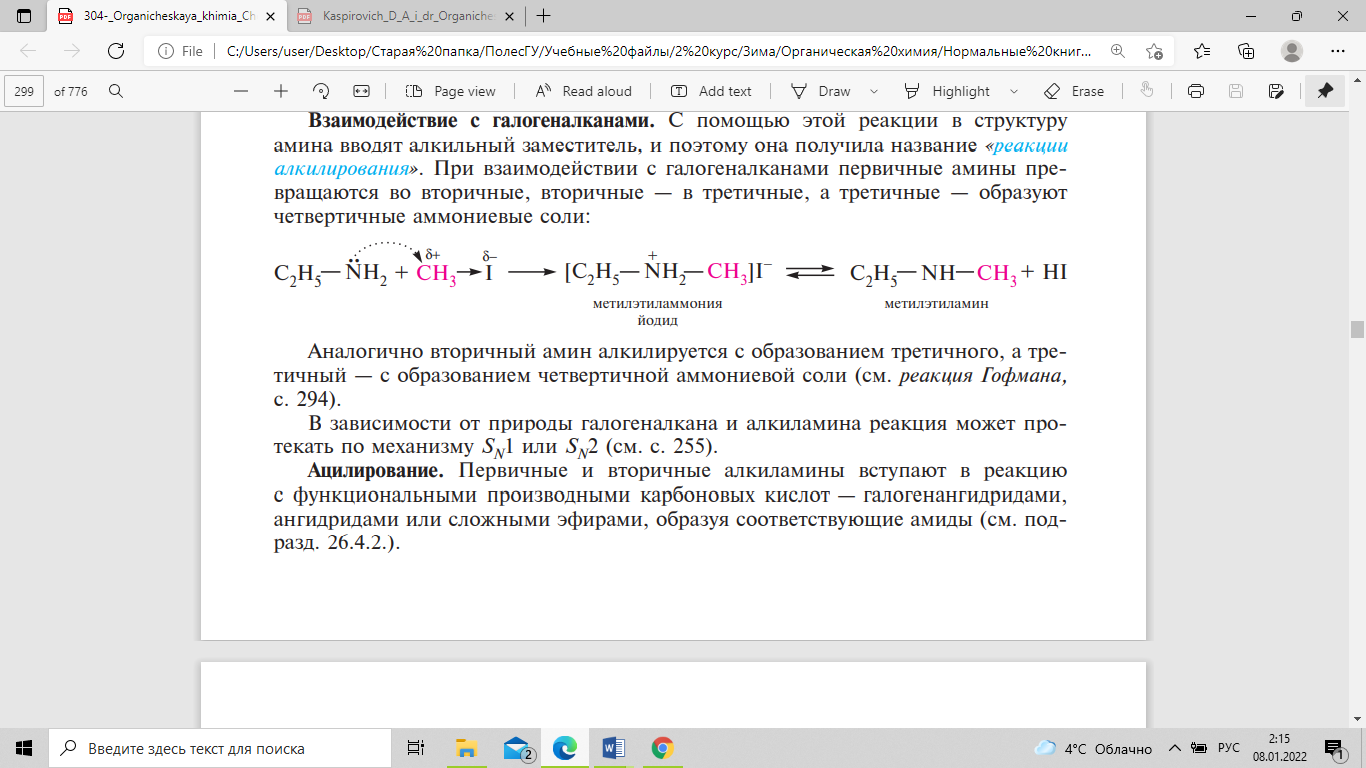
При
действии на аммониевую соль сильного
основания — натрия гидроксида
высвобождается исходный амин. Реакцию
солеобразования с последующим выделением
свободного амина часто используют для
очистки аминов. При взаимодействии с
галогеналканами первичные амины
превращаются во вторичные, вторичные
— в третичные, а третичные — образуют
четвертичные аммониевые соли:
 В
зависимости от природы галогеналкана
и алкиламина реакция может протекать
по механизму SN1 или SN2. Первичные и
вторичные алкиламины вступают в реакцию
с функциональными производными карбоновых
кислот — галогенангидридами, ангидридами
или сложными эфирами, образуя
соответствующие амиды.
В
зависимости от природы галогеналкана
и алкиламина реакция может протекать
по механизму SN1 или SN2. Первичные и
вторичные алкиламины вступают в реакцию
с функциональными производными карбоновых
кислот — галогенангидридами, ангидридами
или сложными эфирами, образуя
соответствующие амиды.
 В
процессе реакции атом водорода при
атоме азота в молекуле амина замещается
на остаток карбоновой кислоты COR,
называемый ацильной группой. Реакции,
с помощью которых в молекулу органического
вещества вводится ацильная группа,
называют реакциями ацилирования.
Третичные амины не содержат при атоме
азота атома водорода и поэтому в реакцию
ацилирования не вступают. Ариламинами
называют
производные аммиака, в молекуле которого
один, два или три атома водорода замещены
остатками ароматических углеводородов.
За счет наличия неподеленной пары
электронов на атоме азота ариламины,
как и алкиламины, проявляют основные.
Однако основность ариламинов значительно
ниже основности алкиламинов. Значительное
снижение основности ариламинов
обусловлено сопряжением неподелённой
пары электронов атома азота с π-электронной
системой ароматического ядра. В результате
сопряжения неподеленная пара электронов
частично делокализуется по ароматическому
ядру, и поэтому она становится менее
доступной для координации с протоном.
Подобно алкиламинам, первичные и
вторичные ариламины реагируют с
галогеналканами, образуя N-алкил- и
N,N-диалкилариламины. Ввиду снижения
нуклеофильных свойств атома азота
алкилирование
ариламинов протекает труднее:
В
процессе реакции атом водорода при
атоме азота в молекуле амина замещается
на остаток карбоновой кислоты COR,
называемый ацильной группой. Реакции,
с помощью которых в молекулу органического
вещества вводится ацильная группа,
называют реакциями ацилирования.
Третичные амины не содержат при атоме
азота атома водорода и поэтому в реакцию
ацилирования не вступают. Ариламинами
называют
производные аммиака, в молекуле которого
один, два или три атома водорода замещены
остатками ароматических углеводородов.
За счет наличия неподеленной пары
электронов на атоме азота ариламины,
как и алкиламины, проявляют основные.
Однако основность ариламинов значительно
ниже основности алкиламинов. Значительное
снижение основности ариламинов
обусловлено сопряжением неподелённой
пары электронов атома азота с π-электронной
системой ароматического ядра. В результате
сопряжения неподеленная пара электронов
частично делокализуется по ароматическому
ядру, и поэтому она становится менее
доступной для координации с протоном.
Подобно алкиламинам, первичные и
вторичные ариламины реагируют с
галогеналканами, образуя N-алкил- и
N,N-диалкилариламины. Ввиду снижения
нуклеофильных свойств атома азота
алкилирование
ариламинов протекает труднее:
 Используется
для получения смешанных аминов. При
действии на первичные и вторичные
ариламины галогенангидридов или
ангидридов карбоновых кислот атомы
водорода аминогруппы замещаются на
ацильные остатки
В
результате реакции образуются замещенные
амиды карбоновых кислот. N-Ацильные
производные анилина и его гомологов
называют анилидами:
В
N-ацильных производных ароматических
аминов неподеленная пара электронов
атома азота находится в сопряжении не
только с π-электронной системой
бензольного ядра, но и с π-электронами
двойной связи карбонильной группы C=O,
что приводит к значительному снижению
в сравнении с аминами ее электронодонорных
свойств по отношению к бензольному
кольцу.
Используется
для получения смешанных аминов. При
действии на первичные и вторичные
ариламины галогенангидридов или
ангидридов карбоновых кислот атомы
водорода аминогруппы замещаются на
ацильные остатки
В
результате реакции образуются замещенные
амиды карбоновых кислот. N-Ацильные
производные анилина и его гомологов
называют анилидами:
В
N-ацильных производных ароматических
аминов неподеленная пара электронов
атома азота находится в сопряжении не
только с π-электронной системой
бензольного ядра, но и с π-электронами
двойной связи карбонильной группы C=O,
что приводит к значительному снижению
в сравнении с аминами ее электронодонорных
свойств по отношению к бензольному
кольцу.
56. Особенности свойств ариламинов. Реакции электрофильного замещения в бензольном ядре ариламинов и их производных. Реакции диазотирования, соли арилдиазония. Реакции солей арилдиазония с выделением азота (замещение диазогруппы) и без выделения азота (азосочетание). Азокрасители.
Ответ.
Ариламинами
называют производные аммиака, в молекуле
которого один, два или три атома водорода
замещены остатками ароматических
углеводородов. Для ариламинов характерны
реакции с участием аминогруппы и реакции
с участием атомов углерода ароматического
ядра. За счет наличия неподеленной пары
электронов на атоме азота проявляют
основные свойства. Однако основность
ариламинов значительно ниже основности
алкиламинов, что обусловлено сопряжением
неподелённой пары электронов атома
азота с π-электронной системой
ароматического ядра. В результате
сопряжения неподеленная пара электронов
частично делокализуется по ароматическому
ядру, и поэтому она становится менее
доступной для координации с протоном.
На основность ариламинов существенное
влияние оказывают заместители в
бензольном кольце. Электронодонорные
заместители увеличивают основность, а
электроноакцепторные — уменьшают ее.
Основность сильно снижается при переходе
от первичных к третичным. Ариламины
образуют соли только с сильными
минеральными кислотами. Для ариламинов
характерны реакции электрофильного
замещения по ароматическому ядру,
свойственные ароматическим углеводородам.
Аминогруппа в молекуле ариламина в
результате проявления +М-эффекта
выступает в качестве сильного
электронодонорного заместителя по
отношению к бензольному кольцу и тем
самым активизирует его реакционную
способность в реакциях электрофильного
замещения. Поэтому ариламины вступают
в реакции электрофильного замещения
значительно легче, чем бензол. Аминогруппа,
являясь заместителем I рода, направляет
электрофильное замещение в орто- и
пара-положения. Анилин легко реагирует
с галогенами (Cl2, Br2) в отсутствие
катализатора, образуя 2,4,6-тригалогенопроизводные
ариламины. Так, при обработке анилина
бромной водой практически с количественным
выходом образуется осадок
2,4,6-триброманилина:
 Хлор
в водных растворах окисляет анилин,
поэтому хлорирование с образованием
2,4,6-трихлоранилина осуществляют действием
хлора в неводных растворителях. При
получении моногалогенозамещенных
ариламинов амины первоначально переводят
в N-ацильные производные, которые затем
галогенируют и гидролизуют.
N-Ацетиламиногруппа является заместителем
I рода, но ее активирующее влияние на
бензольное кольцо значительно меньше,
чем аминогруппы, так как неподеленная
пара электронов атома азота участвует
в сопряжении не только с π-электронной
системой бензольного ядра, но и с
π-электронами двойной связи карбонильной
группы:
Хлор
в водных растворах окисляет анилин,
поэтому хлорирование с образованием
2,4,6-трихлоранилина осуществляют действием
хлора в неводных растворителях. При
получении моногалогенозамещенных
ариламинов амины первоначально переводят
в N-ацильные производные, которые затем
галогенируют и гидролизуют.
N-Ацетиламиногруппа является заместителем
I рода, но ее активирующее влияние на
бензольное кольцо значительно меньше,
чем аминогруппы, так как неподеленная
пара электронов атома азота участвует
в сопряжении не только с π-электронной
системой бензольного ядра, но и с
π-электронами двойной связи карбонильной
группы:
 Нитрование
ариламинов в отличие от аренов имеет
ряд особенностей. Прямое нитрование
ароматических аминов концентрированной
азотной кислотой осуществить невозможно,
так как они легко окисляются. При
использовании в качестве нитрующего
реагента нитрующей смеси ариламины,
наряду с частично протекающими
окислительными процессами, превращаются
в ариламмонийные соли. Аммонийная
группа, являясь электроноакцепторным
заместителем, затрудняет нитрование и
способствует образованию преимущественно
мета-изомера:
С
целью защиты аминогруппы от процессов
окисления и протонирования по атому
азота ароматические амины предварительно
ацетилируют. N-Ацетильные производные
в отличие от аминов являются очень
слабыми основаниями и даже в сильнокислой
среде реагируют в непротонированной
форме. Вместе с тем N-ацетиламиногруппа
сохраняет электронодонорные свойства
и ориентирует нитрование в орто- и
пара-положения. Соотношение орто- и
пара-изомеров зависит от состава
нитрующей смеси и условий проведения
реакции. После нитрования N-ацильные
производные гидролизуют в кислой или
щелочной среде. При нагревании анилина
с концентрированной серной кислотой в
среде высококипящего растворителя
образуется n-аминобензолсульфокислота,
которую чаще называют сульфаниловой
кислотой. Реакция протекает через стадию
образования N-фенилсульфаминовой
кислоты, которая перегруппировывается
в n-аминобензолсульфокислоту:
Нитрование
ариламинов в отличие от аренов имеет
ряд особенностей. Прямое нитрование
ароматических аминов концентрированной
азотной кислотой осуществить невозможно,
так как они легко окисляются. При
использовании в качестве нитрующего
реагента нитрующей смеси ариламины,
наряду с частично протекающими
окислительными процессами, превращаются
в ариламмонийные соли. Аммонийная
группа, являясь электроноакцепторным
заместителем, затрудняет нитрование и
способствует образованию преимущественно
мета-изомера:
С
целью защиты аминогруппы от процессов
окисления и протонирования по атому
азота ароматические амины предварительно
ацетилируют. N-Ацетильные производные
в отличие от аминов являются очень
слабыми основаниями и даже в сильнокислой
среде реагируют в непротонированной
форме. Вместе с тем N-ацетиламиногруппа
сохраняет электронодонорные свойства
и ориентирует нитрование в орто- и
пара-положения. Соотношение орто- и
пара-изомеров зависит от состава
нитрующей смеси и условий проведения
реакции. После нитрования N-ацильные
производные гидролизуют в кислой или
щелочной среде. При нагревании анилина
с концентрированной серной кислотой в
среде высококипящего растворителя
образуется n-аминобензолсульфокислота,
которую чаще называют сульфаниловой
кислотой. Реакция протекает через стадию
образования N-фенилсульфаминовой
кислоты, которая перегруппировывается
в n-аминобензолсульфокислоту:
 Сульфаниловая
кислота является структурным фрагментом
большой группы ЛП — сульфаниламидов.
Соли
арендиазония
образуются при взаимодействии первичных
ароматических аминов с азотистой
кислотой. В промышленности соли
арендиазония нашли широкое применение
для получения разнообразных азокрасителей
всех цветов и оттенков. По этой причине
диазотирование относится к числу
важнейших и наиболее изученных реакций
в органической химии.
Диазотирование
первичных ароматических аминов
описывается следующим суммарным
уравнением:
ArNH2+
NaNO2+
2 HCl
Сульфаниловая
кислота является структурным фрагментом
большой группы ЛП — сульфаниламидов.
Соли
арендиазония
образуются при взаимодействии первичных
ароматических аминов с азотистой
кислотой. В промышленности соли
арендиазония нашли широкое применение
для получения разнообразных азокрасителей
всех цветов и оттенков. По этой причине
диазотирование относится к числу
важнейших и наиболее изученных реакций
в органической химии.
Диазотирование
первичных ароматических аминов
описывается следующим суммарным
уравнением:
ArNH2+
NaNO2+
2 HCl![]() ArN+=N
Cl - +
NaCl + 2 H2O
Реакции
с выделением
азота.
При кипячении кислых растворов солей
диазония происходит выделение азота и
получаются фенолы. Превращение солей
диазония без выделения азота. Реакции
этой группы делают возможным переход
от диазосоединений к азосоединениям
(производным азобензола). Органические
вещества этого класса лежат в основе
одного из разделов промышленности,
производящей синтетические красители
из продуктов, добываемых из каменноугольного
дегтя. Все азокрасители
получаются при помощи так называемой
реакции сочетания солей диазония
— химическая реакция, в ходе которой к
ароматическому диазосоединению
присоединяется другое соединение,
называемое азосоставляющим, содержащее
способный к замещению атом водорода,
либо в некоторых случаях, другие атомы
или группы, в результате чего происходит
образование азосоединения. Диазосоединение
при вступлении в реакцию азосочетания
находится в форме катиона диазония.
Сама реакция имеет две ступени, из
которых медленной является первая —
присоединение катиона к азосоставляющей.
Вторая стадия — отщепление протона,
которую можно ускорить при помощи
введения в реакционную массу акцепторов
протонов, в роли которых используют
карбонатные и ацетатные группы. Пример
реакции азосочетания — получение
4-гидроксиазобензола:
ArN+=N
Cl - +
NaCl + 2 H2O
Реакции
с выделением
азота.
При кипячении кислых растворов солей
диазония происходит выделение азота и
получаются фенолы. Превращение солей
диазония без выделения азота. Реакции
этой группы делают возможным переход
от диазосоединений к азосоединениям
(производным азобензола). Органические
вещества этого класса лежат в основе
одного из разделов промышленности,
производящей синтетические красители
из продуктов, добываемых из каменноугольного
дегтя. Все азокрасители
получаются при помощи так называемой
реакции сочетания солей диазония
— химическая реакция, в ходе которой к
ароматическому диазосоединению
присоединяется другое соединение,
называемое азосоставляющим, содержащее
способный к замещению атом водорода,
либо в некоторых случаях, другие атомы
или группы, в результате чего происходит
образование азосоединения. Диазосоединение
при вступлении в реакцию азосочетания
находится в форме катиона диазония.
Сама реакция имеет две ступени, из
которых медленной является первая —
присоединение катиона к азосоставляющей.
Вторая стадия — отщепление протона,
которую можно ускорить при помощи
введения в реакционную массу акцепторов
протонов, в роли которых используют
карбонатные и ацетатные группы. Пример
реакции азосочетания — получение
4-гидроксиазобензола:
 Реакцию
азосочетания обычно проводят при
температуре от 0 до 25 °C, повышая её при
необходимости до 40—50 °C в случаях
малоактивных диазосоединений, так как
при этом последние разрушаются с
выделением азота.
Реакцию
азосочетания обычно проводят при
температуре от 0 до 25 °C, повышая её при
необходимости до 40—50 °C в случаях
малоактивных диазосоединений, так как
при этом последние разрушаются с
выделением азота.