

18. Представление о стереохимии присоединения галогенов у алкенов. Перегруппировки карбокатионов. Реакции радикального присоединения (по Харашу).
Ответ. Алкены реагируют с бромом и хлором с образованием продуктов присоединения по двойной связи одной молекулы галогена с выходом близким к количественному. Фтор слишком активен и вызывает деструкцию алкенов. Присоединение йода к алкенам в большинстве случаев представляет собой обратимую реакцию, равновесие которой смещено в сторону исходных реагентов.
 Присоединение
брома и хлора к алкенам происходит по
ионному, а не по радикальному механизму.
Этот вывод следует из того, что скорость
присоединения галогена не зависит от
облучения, присутствия кислорода и
других реагентов, инициирующих или
ингибирующих радикальные процессы. На
основании большого числа экспериментальных
данных для этой реакции был предложен
механизм, включающий несколько
последовательных стадий. На первой
стадии происходит поляризация молекулы
галогена под действием электронов
π-связи. Атом галогена, приобретающий
некоторый дробный положительный заряд,
образует с электронами π-связи нестабильный
интермедиат, называемый -комплексом
или комплексом с переносом заряда.
Следует отметить, что в π-комплексе
галоген не образует направленной связи
с каким-нибудь конкретным атомом
углерода; в этом комплексе просто
реализуется донорно-акцепторное
взаимодействие электронной пары π-связи
как донора и галогена как акцептора.
Далее π-комплекс превращается в
циклический бромониевый ион. В процессе
образования этого циклического катиона
происходит гетеролитический разрыв
связи Br-Br и пустая р-орбиталь
sp2-гибридизованного атома углерода
перекрывается с р-орбиталью "неподеленной
пары" электронов атома галогена,
образуя циклический ион бромония.
Присоединение галогенов к двойной связи
алкенов представляет собой одну из
формально простых модельных реакций,
на примере которой можно рассмотреть
влияние основных факторов, позволяющих
сделать аргументированные выводы о
детальном механизме процесса. Для
обоснованных выводов о механизме любой
реакции следует располагать данными
по: 1) кинетике реакции; 2) стереохимии
(стереохимический результат реакции);
3) наличию или отсутствию сопряженного,
конкурирующего процесса; 4) влиянию
заместителей в исходном субстрате на
скорость реакции; 5) использованию
меченых субстратов и (или) реагентов;
6) возможности перегруппировок в ходе
реакции; 7) влиянию растворителя на
скорость реакции. Наиболее ценную
информацию о механизме этой реакции
представляют данные по стереохимии
присоединения. Присоединение галогена
к двойной связи представляет собой
стереоспецифический процесс (процесс,
в котором образуется только один из
возможных стереоизомеров; в стереоселективном
процессе наблюдается преимущественное
образование одного стереомера)
анти-присоединения для алкенов и
циклоалкенов, у которых двойная связь
не сопряжена с бензольным кольцом. Для
цис- и транс-изомеров бутена-2, пентена-2,
гексена-3, циклогексена, циклопентена
и других алкенов присоединение брома
происходит исключительно как
анти-присоединение. При этом в случае
циклогексена образуется исключительно
транс-1,2-дибромциклогексан (смесь
энантиомеров).
Присоединение
брома и хлора к алкенам происходит по
ионному, а не по радикальному механизму.
Этот вывод следует из того, что скорость
присоединения галогена не зависит от
облучения, присутствия кислорода и
других реагентов, инициирующих или
ингибирующих радикальные процессы. На
основании большого числа экспериментальных
данных для этой реакции был предложен
механизм, включающий несколько
последовательных стадий. На первой
стадии происходит поляризация молекулы
галогена под действием электронов
π-связи. Атом галогена, приобретающий
некоторый дробный положительный заряд,
образует с электронами π-связи нестабильный
интермедиат, называемый -комплексом
или комплексом с переносом заряда.
Следует отметить, что в π-комплексе
галоген не образует направленной связи
с каким-нибудь конкретным атомом
углерода; в этом комплексе просто
реализуется донорно-акцепторное
взаимодействие электронной пары π-связи
как донора и галогена как акцептора.
Далее π-комплекс превращается в
циклический бромониевый ион. В процессе
образования этого циклического катиона
происходит гетеролитический разрыв
связи Br-Br и пустая р-орбиталь
sp2-гибридизованного атома углерода
перекрывается с р-орбиталью "неподеленной
пары" электронов атома галогена,
образуя циклический ион бромония.
Присоединение галогенов к двойной связи
алкенов представляет собой одну из
формально простых модельных реакций,
на примере которой можно рассмотреть
влияние основных факторов, позволяющих
сделать аргументированные выводы о
детальном механизме процесса. Для
обоснованных выводов о механизме любой
реакции следует располагать данными
по: 1) кинетике реакции; 2) стереохимии
(стереохимический результат реакции);
3) наличию или отсутствию сопряженного,
конкурирующего процесса; 4) влиянию
заместителей в исходном субстрате на
скорость реакции; 5) использованию
меченых субстратов и (или) реагентов;
6) возможности перегруппировок в ходе
реакции; 7) влиянию растворителя на
скорость реакции. Наиболее ценную
информацию о механизме этой реакции
представляют данные по стереохимии
присоединения. Присоединение галогена
к двойной связи представляет собой
стереоспецифический процесс (процесс,
в котором образуется только один из
возможных стереоизомеров; в стереоселективном
процессе наблюдается преимущественное
образование одного стереомера)
анти-присоединения для алкенов и
циклоалкенов, у которых двойная связь
не сопряжена с бензольным кольцом. Для
цис- и транс-изомеров бутена-2, пентена-2,
гексена-3, циклогексена, циклопентена
и других алкенов присоединение брома
происходит исключительно как
анти-присоединение. При этом в случае
циклогексена образуется исключительно
транс-1,2-дибромциклогексан (смесь
энантиомеров).
 При
присоединении брома к циклогексену
первоначально образуется
транс-1,2-дибромциклогексан в а,а-конформации,
которая затем сразу же переходит в
энергетически более выгодную
е,е-конформацию. Анти-присоединение
галогенов к двойной связи позволяет
отвергнуть механизм одностадийного
синхронного присоединения одной молекулы
галогена к двойной связи, которое может
осуществляться только как син-присоединение.
Анти-присоединение галогена не согласуется
также и с образованием открытого
карбкатиона RCH+-CH2Hal в качестве интермедиата.
В открытом карбокатионе возможно
свободное вращение вокруг С-С-связи,
что должно приводить после атаки аниона
Br- к образованию смеси продуктов как
анти-, так и син-присоединения.
Стереоспецифическое анти-присоединение
галогенов явилось главной причиной
создания концепции бромониевого или
хлорониевого ионов в качестве дискретных
промежуточных частиц. Эта концепция
идеально удовлетворяет правилу
анти-присоединения, поскольку нуклеофильная
атака галогенид-иона возможна с
анти-стороны по любому из двух атомов
углерода галогенониевого иона по SN2
механизму. В некоторых случаях
присоединение хлора к алкенам, содержащим
электронодонорные заместители,
сопровождается отщеплением протона из
промежуточного соединения вместо
присоединения хлорид-иона. Отщепление
протона приводит к образованию
хлорзамещенного алкена, которое формально
можно рассматривать как прямое замещение
с миграцией двойной связи. Однако опыты
с изотопной меткой указывают на более
сложный характер происходящих здесь
превращений. При хлорировании изобутилена
при 0°С образуется 2-метил-3-хлорпропен
(металлилхлорид) вместо ожидаемого
дихлорида - продукта присоединения по
двойной связи. Скелетные перегруппировки
подобного типа наиболее характерны для
процессов с участием открытых карбокатионов
в качестве промежуточных частиц. Не
исключено, что присоединение хлора в
этих случаях идет не через хлорониевый
ион, а через катионную частицу, близкую
к открытому карбокатиону. Вместе с тем
следует отметить, что скелетные
перегруппировки явление достаточно
редкое в процессах присоединения
галогенов и смешанных галогенов по
двойной связи: они чаще наблюдаются при
присоединении хлора и гораздо реже при
присоединении брома. Вероятность таких
перегруппировок увеличивается при
переходе от неполярных растворителей
(ССl4) к полярным (нитрометан, ацетонитрил).
При
присоединении брома к циклогексену
первоначально образуется
транс-1,2-дибромциклогексан в а,а-конформации,
которая затем сразу же переходит в
энергетически более выгодную
е,е-конформацию. Анти-присоединение
галогенов к двойной связи позволяет
отвергнуть механизм одностадийного
синхронного присоединения одной молекулы
галогена к двойной связи, которое может
осуществляться только как син-присоединение.
Анти-присоединение галогена не согласуется
также и с образованием открытого
карбкатиона RCH+-CH2Hal в качестве интермедиата.
В открытом карбокатионе возможно
свободное вращение вокруг С-С-связи,
что должно приводить после атаки аниона
Br- к образованию смеси продуктов как
анти-, так и син-присоединения.
Стереоспецифическое анти-присоединение
галогенов явилось главной причиной
создания концепции бромониевого или
хлорониевого ионов в качестве дискретных
промежуточных частиц. Эта концепция
идеально удовлетворяет правилу
анти-присоединения, поскольку нуклеофильная
атака галогенид-иона возможна с
анти-стороны по любому из двух атомов
углерода галогенониевого иона по SN2
механизму. В некоторых случаях
присоединение хлора к алкенам, содержащим
электронодонорные заместители,
сопровождается отщеплением протона из
промежуточного соединения вместо
присоединения хлорид-иона. Отщепление
протона приводит к образованию
хлорзамещенного алкена, которое формально
можно рассматривать как прямое замещение
с миграцией двойной связи. Однако опыты
с изотопной меткой указывают на более
сложный характер происходящих здесь
превращений. При хлорировании изобутилена
при 0°С образуется 2-метил-3-хлорпропен
(металлилхлорид) вместо ожидаемого
дихлорида - продукта присоединения по
двойной связи. Скелетные перегруппировки
подобного типа наиболее характерны для
процессов с участием открытых карбокатионов
в качестве промежуточных частиц. Не
исключено, что присоединение хлора в
этих случаях идет не через хлорониевый
ион, а через катионную частицу, близкую
к открытому карбокатиону. Вместе с тем
следует отметить, что скелетные
перегруппировки явление достаточно
редкое в процессах присоединения
галогенов и смешанных галогенов по
двойной связи: они чаще наблюдаются при
присоединении хлора и гораздо реже при
присоединении брома. Вероятность таких
перегруппировок увеличивается при
переходе от неполярных растворителей
(ССl4) к полярным (нитрометан, ацетонитрил).
 Присоединение
галогенов к алкенам можно легко
осуществить в газовой фазе и по
радикальному типу при освещении
УФ-светом. Наряду с галогенами по
радикальному типу удается присоединить
к алкенам и галогеноводороды. Как показал
Хараш,
в присутствии перекисей присоединение
бромистого водорода к алкенам идет
против правила Марковникова.
Механизм
реакции радикального присоединения
бромистого водорода к алкенам по Харашу
следующий:
Присоединение
галогенов к алкенам можно легко
осуществить в газовой фазе и по
радикальному типу при освещении
УФ-светом. Наряду с галогенами по
радикальному типу удается присоединить
к алкенам и галогеноводороды. Как показал
Хараш,
в присутствии перекисей присоединение
бромистого водорода к алкенам идет
против правила Марковникова.
Механизм
реакции радикального присоединения
бромистого водорода к алкенам по Харашу
следующий:
 Из
двух возможных радикалов А и Б в основном
образуется первый, как более устойчивый.
Таким образом, по аналогии с электрофильным
радикальное присоединение к алкенам
протекает через стадию образования
наиболее стабильного радикала. Реакция
Хараша характерна для НВr, идет с трудом
с НCl и неизвестна для HF и HI . Если для HF
присоединение термодинамически
неблагоприятно, то для HI — наоборот.
Тем не менее реакцию присоединения HI
не удается осуществить, поскольку легко
идет обратный процесс восстановления
йодистым водородом алкилиодида.
Из
двух возможных радикалов А и Б в основном
образуется первый, как более устойчивый.
Таким образом, по аналогии с электрофильным
радикальное присоединение к алкенам
протекает через стадию образования
наиболее стабильного радикала. Реакция
Хараша характерна для НВr, идет с трудом
с НCl и неизвестна для HF и HI . Если для HF
присоединение термодинамически
неблагоприятно, то для HI — наоборот.
Тем не менее реакцию присоединения HI
не удается осуществить, поскольку легко
идет обратный процесс восстановления
йодистым водородом алкилиодида.
19. Реакции окисления алкенов по С=С связи: цис-дигидроксилирование (перманганатом калия по Вагнеру, тетраоксидом осмия), эпоксидирование (по Прилежаеву).
Ответ.
Алкены окисляются довольно легко.
Направление окисления зависит от природы
окислителя и условий проведения реакции.
Разбавленный раствор калия
перманганата
в нейтральной или слабощелочной среде
окисляет алкены до двухатомных спиртов
(гликолей). При этом обесцвечивается
розово-фиолетовая окраска раствора
калия перманганата и выпадает коричневый
осадок марганца (IV) оксида:
 Данная
реакция была открыта в 1888 году русским
химиком Вагнером. Реакция Вагнера
используется в фармацевтическом анализе
для обнаружения двойной углерод-углеродной
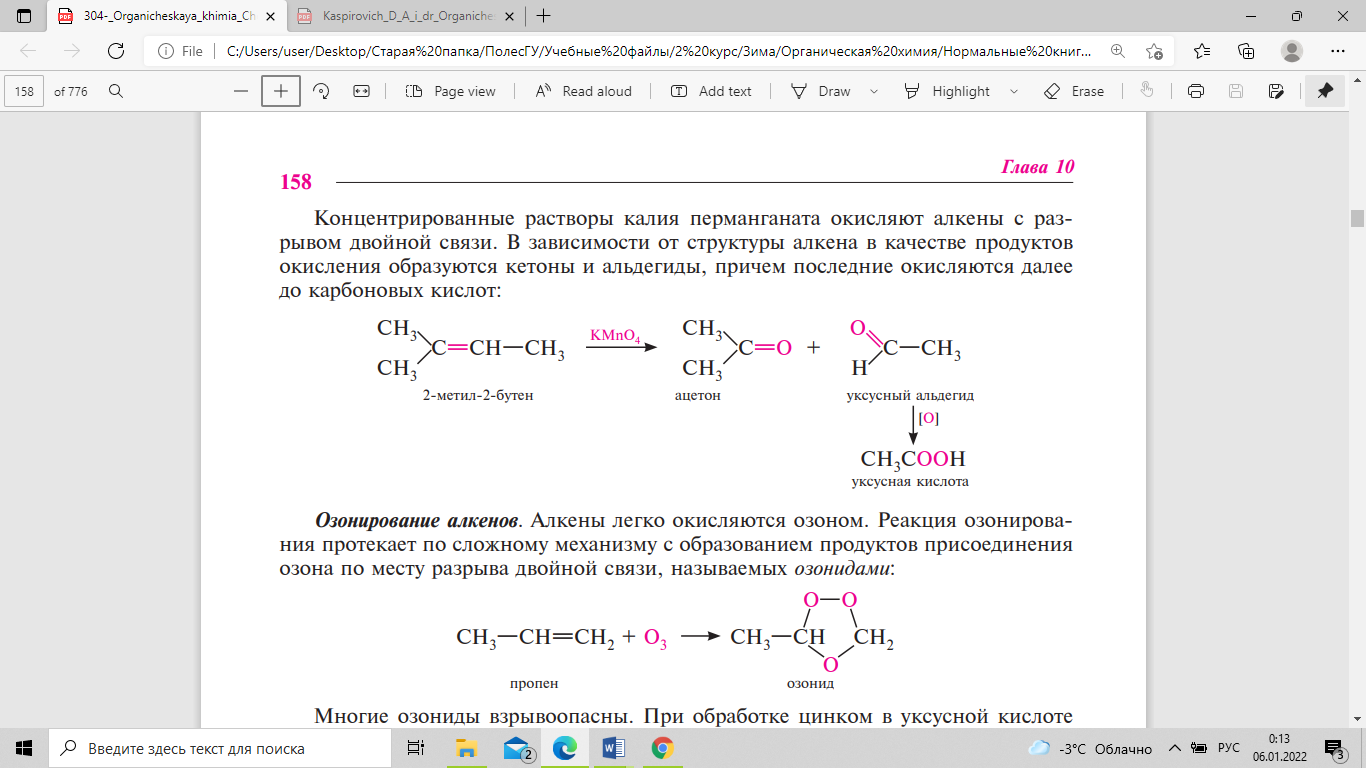
связи. Концентрированные растворы калия
перманганата окисляют алкены с разрывом
двойной связи. В зависимости от структуры
алкена в качестве продуктов окисления
образуются кетоны и альдегиды, причем
последние окисляются далее до карбоновых
кислот:
Данная
реакция была открыта в 1888 году русским
химиком Вагнером. Реакция Вагнера
используется в фармацевтическом анализе
для обнаружения двойной углерод-углеродной
связи. Концентрированные растворы калия
перманганата окисляют алкены с разрывом
двойной связи. В зависимости от структуры
алкена в качестве продуктов окисления
образуются кетоны и альдегиды, причем
последние окисляются далее до карбоновых
кислот:
 Другой
метод гидроксилирования алкенов под
действием оксида осмия (VIII) OsO4 был
предложен Р. Криге в 1936 году. Тетраоксид
осмия представляет собой бесцветное,
летучее, кристаллическое вещество,
хорошо растворимое в органических
растворителях. При взаимодействии
тетраоксида осмия с алкенами образуется
черный осадок циклического эфира
осмиевой кислоты - осмат, который легко
может быть изолирован в индивидуальном
виде. Присоединение OsO4 к двойной связи
заметно ускоряется в растворе в пиридине.
Разложение осматов до вицинальных
гликолей достигается действием водного
раствора гидросульфита натрия или
сероводородом.
Другой
метод гидроксилирования алкенов под
действием оксида осмия (VIII) OsO4 был
предложен Р. Криге в 1936 году. Тетраоксид
осмия представляет собой бесцветное,
летучее, кристаллическое вещество,
хорошо растворимое в органических
растворителях. При взаимодействии
тетраоксида осмия с алкенами образуется
черный осадок циклического эфира
осмиевой кислоты - осмат, который легко
может быть изолирован в индивидуальном
виде. Присоединение OsO4 к двойной связи
заметно ускоряется в растворе в пиридине.
Разложение осматов до вицинальных
гликолей достигается действием водного
раствора гидросульфита натрия или
сероводородом. Выходы
продуктов в этом методе значительно
выше, чем при использовании перманганата
в качестве окислителя. Важным достоинством
метода Криге является отсутствие
продуктов окислительного расщепления
алкенов, характерного для перманганатного
окисления. Тетраоксид осмия очень
дорогой и труднодоступный реагент, к
тому же он токсичен. Кислород воздуха
в присутствии серебряного катализатора
окисляет алкены при нагревании с
образованием эпоксидов:
Выходы
продуктов в этом методе значительно
выше, чем при использовании перманганата
в качестве окислителя. Важным достоинством
метода Криге является отсутствие
продуктов окислительного расщепления
алкенов, характерного для перманганатного
окисления. Тетраоксид осмия очень
дорогой и труднодоступный реагент, к
тому же он токсичен. Кислород воздуха
в присутствии серебряного катализатора
окисляет алкены при нагревании с
образованием эпоксидов:
 Реакция
применяется в промышленности для
получения этиленоксида (оксирана).
Аналогично алкены окисляются
пероксикислотами (реакция Прилежаева).
Так, при обработке алкенов пероксибензойной
кислотой образуются эпоксиды:
Реакция
применяется в промышленности для
получения этиленоксида (оксирана).
Аналогично алкены окисляются
пероксикислотами (реакция Прилежаева).
Так, при обработке алкенов пероксибензойной
кислотой образуются эпоксиды:
 Реакция
изучена в 1909 году российским
химиком-органиком Н. А. Прилежаевым.
Реакция
изучена в 1909 году российским
химиком-органиком Н. А. Прилежаевым.