

14. Химические свойства алканов. Общие представления о механизме цепных радикальных реакций замещения в алканах (на примере реакции галогенирования).
Ответ.
В обычных условиях алканы являются
малореакционноспособными соединениями.
Они устойчивы к действию кислот, щелочей
и окислителей. Инертность алканов
используют на практике. Концентрированную
серную кислоту и щелочи применяют для
очистки нефтепродуктов. Щелочные
металлы, для предотвращения контакта
с кислородом и влагой воздуха, хранят
под слоем инертного растворителя
(керосин). Химическая инертность алканов
обусловлена высокой прочностью σ-связей
С—С и С—Н. В результате незначительного
отличия электроотрицательностей
sp3-гибридизованного атома углерода и
атома водорода σ-связи С—С и С—Н в
алканах практически не полярны и поэтому
не склонны к гетеролитическому разрыву,
но способны расщепляться гомолитически
с образованием свободных радикалов.
Несмотря на то что связь С—С менее
прочная, чем связь С—Н, последняя
разрывается с большей легкостью, так
как более доступна для атаки реагентом.
Химические превращения алканов
сопровождаются гомолитическим
расщеплением связей С—Н с последующим
замещением атома водорода другими
атомами или группами, то есть для них
характерны реакции замещения, происходящие
по радикальному механизму (SR). При высоких
температурах может наблюдаться
гомолитический разрыв связей С—С.
Алканы легко реагируют с галогенами
(кроме йода), образуя смеси моно- и
полигалогеналканов. Галогены по
реакционной способности с алканами
располагаются в ряд: F2 > Сl2 > Вr2. Прямое
фторирование алканов является трудно
контролируемой экзотермической реакцией.
Выделяющаяся энергия при замещении
атома водорода на атом фтора превышает
энергию диссоциации связи С—С. При
фторировании алканов наряду с замещением
атомов водорода атомами фтора происходит
разрыв углерод-углеродных связей, и
образуется сложная смесь фторалканов.
Именно поэтому прямое фторирование
алканов имеет ограниченное применение.
Менее экзотермична реакция алканов с
хлором. Она протекает в условиях
фотохимического (под действием
УФ-облучения) или термического (300 °С)
процесса. При взаимодействии метана с
хлором атомы водорода постепенно
замещаются на атомы хлора:
 .
Реакция протекает по цепному
свободнорадикальному механизму, который
.
Реакция протекает по цепному
свободнорадикальному механизму, который
был
изучен советским ученым Николаем
Николаевичем Семеновым. В цепном процессе
выделяют три стадии: инициирование,
рост цепи, обрыв цепи. Инициирование.
Под действием энергии квантов света
(hn) или нагревания молекула хлора
активируется и претерпевает гомолитический
разрыв связи с образованием двух
свободных радикалов: Cl· ·Cl hn 2Cl·. Рост
цепи. Свободные радикалы хлора атакуют
связь С—Н в молекуле метана, отрывая
при этом атом водорода с образованием
хлороводорода HCl и свободного метильного
радикала СН3. Метильный радикал, в свою
очередь, атакует молекулу хлора, отрывает
атом галогена и образует хлорметан
СН3Сl и свободный радикал хлора.
Образовавшийся радикал хлора повторяет
цикл указанных превращений, то есть
происходит цепной процесс, в котором
атом хлора, прореагировавший на предыдущей
стадии роста цепи, способствует
высвобождению нового радикала хлора
на последующей стадии. В результате
образуется смесь моно-, ди-, три- и
тетрахлорпроизводных метана. Цепной
процесс прекращается только после
исчезновения всех свободных радикалов,
образующихся в ходе реакции. Обрыв цепи.
В результате рекомбинации (димеризации)
свободных радикалов происходит обрыв
цепи. Аналогично протекает реакция
бромирования метана, однако она
значительно менее экзотермична, чем
реакции фторирования и хлорирования.
Следует отметить, что замещение атома
водорода на атом галогена в алканах
происходит в большинстве случаев
региоселективно (избирательно): в первую
очередь, как правило, замещается атом
водорода при третичном атоме углерода,
затем — при вторичном и в последнюю
очередь — при первичном. Такая
последовательность замещения обусловлена
устойчивостью образующихся свободных
радикалов. Чем устойчивее свободный
радикал, тем легче он образуется.
Поскольку третичные алкильные радикалы
более стабильны, чем вторичные и тем
более первичные, реакционная способность
связей С—Н при галогенировании алканов
увеличивается в ряду: первичный <
вторичный < третичный атом углерода.
Однако эта закономерность строго не
выполняется. Региоселективность
галогенирования зависит также от
активности реагента (атома галогена) и
температуры. Чем активнее реагент, тем
ниже селективность реакции. Следовательно,
у алканов реакция бромирования более
селективна, чем хлорирования.
Региоселективность галогенирования
алканов возрастает при понижении
температуры. Радикальное йодирование
алканов эндотермично и вследствие
низкой реакционной активности I протекает
с трудом. Реакция обратима, так как
образующийся йодоводород HI восстанавливает
алкилйодид. Под действием серы
(IV) оксида и хлора в условиях УФ-облучения
алканы образуют алкансульфонилхлориды
(хлорангидриды алкансульфоновых кислот):
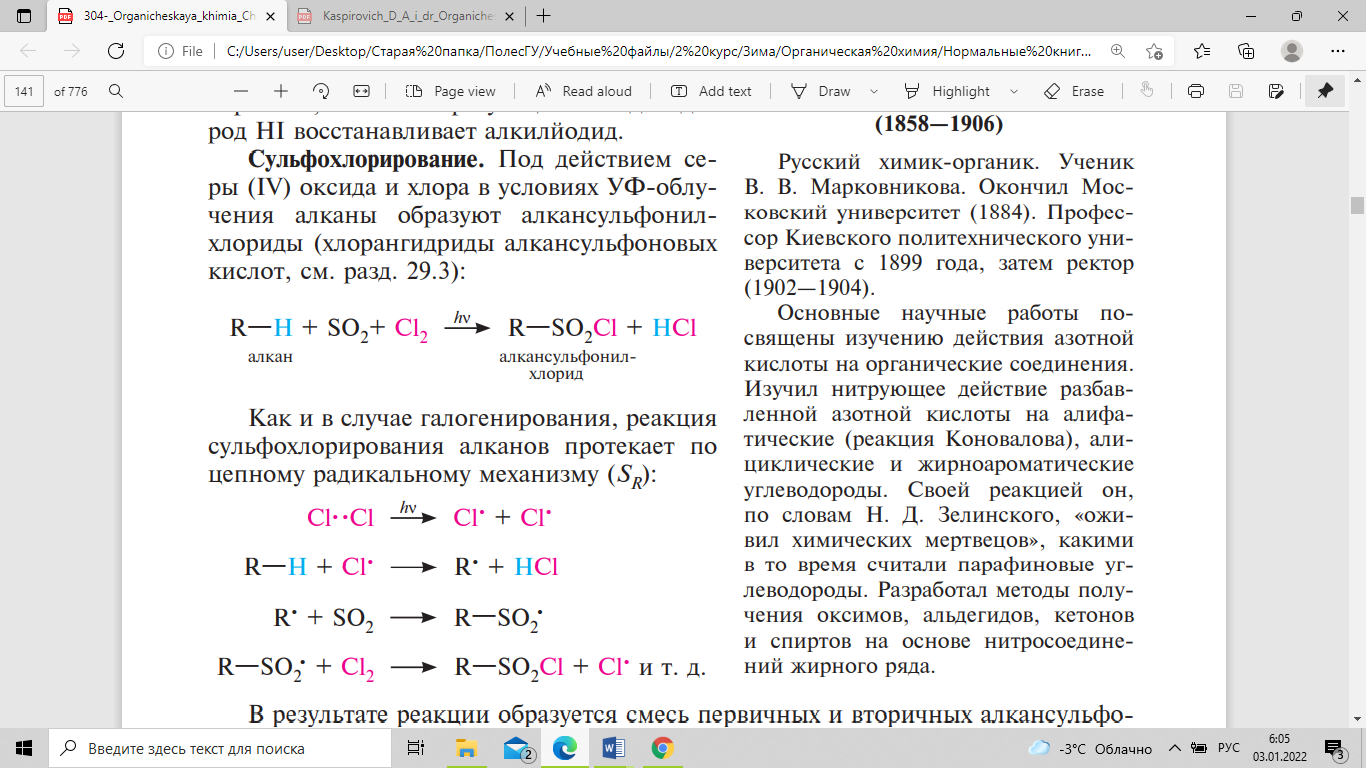 .
Как и в случае галогенирования, реакция
сульфохлорирования алканов протекает
по цепному радикальному механизму (SR):
.
В результате реакции образуется смесь
первичных и вторичных алкансульфонилхлоридов.
Третичные сульфонилхлориды не образуются,
очевидно, вследствие пространственных
препятствий. Реакция сульфохлорирования
имеет важное значение в производстве
СМС. Метод жидкофазного нитрования
алканов, называемый реакцией Коновалова
(1888), осуществляется с использованием
разбавленной азотной кислоты (концентрация
10—20 %) при температуре 110—140 °С, нормальном
или повышенном давлении:
.
Как и в случае галогенирования, реакция
сульфохлорирования алканов протекает
по цепному радикальному механизму (SR):
.
В результате реакции образуется смесь
первичных и вторичных алкансульфонилхлоридов.
Третичные сульфонилхлориды не образуются,
очевидно, вследствие пространственных
препятствий. Реакция сульфохлорирования
имеет важное значение в производстве
СМС. Метод жидкофазного нитрования
алканов, называемый реакцией Коновалова
(1888), осуществляется с использованием
разбавленной азотной кислоты (концентрация
10—20 %) при температуре 110—140 °С, нормальном
или повышенном давлении:
![]()
Реакция
протекает по свободнорадикальному
механизму. Нитрование алканов
характеризуется высокой региоселективностью.
Концентрированная азотная кислота в
обычных условиях не взаимодействует с
алканами; при нагревании она действует
главным образом как окислитель. В избытке
кислорода или на воздухе алканы сгорают
с образованием углерода (IV) оксида и
воды с выделением большого количества
теплоты: СН4 + 2О2 → СO2 + 2Н2O + 882 кДж/моль.
Окисление алканов кислородом воздуха
в присутствии катализатора (солей
марганца, хрома, свинца и др.) при
температуре 150—200 °С приводит к образованию
смеси продуктов, состоящей в основном
из карбоновых кислот с различной длиной
углеродной цепи, альдегидов, кетонов и
спиртов. Реакция протекает по радикальному
механизму и сопровождается разрывом
углерод-углеродных связей. В качестве
промежуточных продуктов окисления
образуются органические гидропероксиды.
 .
Реакция окисления используется в
промышленности для получения метанола,
формальдегида, ацетальдегида и уксусной
кислоты из пропана и бутана, а также
высших жирных кислот из алканов с длиной
цепи более 25 углеродных атомов. Крекинг
— процесс термического расщепления
алканов. Под действием высоких температур
алканы разлагаются с разрывом связей
С—С и С—Н. Одновременно протекают
процессы дегидрирования, изомеризации
и циклизации. Начальная температура
распада алканов зависит от их строения
и молекулярной массы. Чем больше
молекулярная масса углеводорода, тем
легче он расщепляется при нагревании.
Различают термический крекинг и
каталитический крекинг. Термический
крекинг проводят при температуре 800 °С
и выше, каталитический — при температуре
450—550 °С в присутствии алюмосиликатных
катализаторов (алюминия оксид Аl2О3 на
силикагеле SiO2). Наиболее устойчив к
термическому разложению метан. В
интервале температур 1400—1500 °С он
подвергается распаду с образованием
ацетилена: 2СН4 1400—1500 °С НC———СН + 3Н2.
Этан разлагается при более низких
температурах: CH3—CH3 600—800 °С H2C——CH2 + Н2.
Высшие алканы в условиях термического
крекинга разлагаются с образованием
сложной смеси низших алканов и алкенов.
Разрыв углеродной цепи молекулы может
произойти в любом положении. Термический
крекинг протекает по радикальному
механизму. При каталитическом крекинге
расщепление углерод-углеродной связи
сопровождается преимущественно
изомеризацией н-алканов в алканы с
разветвленной цепью. В присутствии
катализатора высшие алканы способны к
циклизации с образованием ароматических
углеводородов. Каталитический крекинг
протекает по ионному механизму.
Крекинг-процесс имеет важное промышленное
значение и широко используется для
получения высокооктановых бензинов,
непредельных и ароматических углеводородов.
.
Реакция окисления используется в
промышленности для получения метанола,
формальдегида, ацетальдегида и уксусной
кислоты из пропана и бутана, а также
высших жирных кислот из алканов с длиной
цепи более 25 углеродных атомов. Крекинг
— процесс термического расщепления
алканов. Под действием высоких температур
алканы разлагаются с разрывом связей
С—С и С—Н. Одновременно протекают
процессы дегидрирования, изомеризации
и циклизации. Начальная температура
распада алканов зависит от их строения
и молекулярной массы. Чем больше
молекулярная масса углеводорода, тем
легче он расщепляется при нагревании.
Различают термический крекинг и
каталитический крекинг. Термический
крекинг проводят при температуре 800 °С
и выше, каталитический — при температуре
450—550 °С в присутствии алюмосиликатных
катализаторов (алюминия оксид Аl2О3 на
силикагеле SiO2). Наиболее устойчив к
термическому разложению метан. В
интервале температур 1400—1500 °С он
подвергается распаду с образованием
ацетилена: 2СН4 1400—1500 °С НC———СН + 3Н2.
Этан разлагается при более низких
температурах: CH3—CH3 600—800 °С H2C——CH2 + Н2.
Высшие алканы в условиях термического
крекинга разлагаются с образованием
сложной смеси низших алканов и алкенов.
Разрыв углеродной цепи молекулы может
произойти в любом положении. Термический
крекинг протекает по радикальному
механизму. При каталитическом крекинге
расщепление углерод-углеродной связи
сопровождается преимущественно
изомеризацией н-алканов в алканы с
разветвленной цепью. В присутствии
катализатора высшие алканы способны к
циклизации с образованием ароматических
углеводородов. Каталитический крекинг
протекает по ионному механизму.
Крекинг-процесс имеет важное промышленное
значение и широко используется для
получения высокооктановых бензинов,
непредельных и ароматических углеводородов.