

ТАКТИКА ВЕДЕНИЯ ДЕТЕЙ С МПС III ТИПА
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС III ТИПА
В настоящее время не существует патогенетического лечения МПС III типа.
Трансплантация стволовых клеток не имеет достаточных данных для широкого применения (описано несколько случаев в США).
Трансплантация костного мозга неэффективна.
Пациенты должны получать симптоматическую терапию в соответствии с выявленными нарушениями и жалобами.
ИСХОДЫ И ПРОГНОЗ
Больные умирают, не достигая возраста 30 лет, часто вследствие возникших инфекций нижних дыхательных путей.
МУКОПОЛИСАХАРИДОЗ IV ТИПА (СИНДРОМ МОРКИО) -
(СИНОНИМЫ: БОЛЕЗНЬ МОРКИО, СПОНДИЛО-ЭПИФИЗАРНАЯ ДИСПЛАЗИЯ, ХОНДРООСТЕОДИСТРОФИЯ, ДЕФОРМИРУЮЩАЯ ОСТЕОХОНДРОДИСТРОФИЯ, МОРКИО - БРАЙЛСФОРДА СИНДРОМ, МОРКИО - УЛЬРИХА СИНДРОМ, К - МУКОПОЛИСАХАРИДОЗ, ЭКСЦЕНТРОХОНДРОПЛАЗИЯ, ДУГВЕ - МЕЛХИОРА - КЛАУЗЕНА СИНДРОМ)
Наследственная болезнь накопления, обусловленная дефицитом лизосомных гидролаз: галактозамин-6-сульфат-сульфатазы (МПС IVА) или β-галактозидазы (МПС IVВ), обусловлена отложением в соединительной ткани кератансульфата и характеризуется значительной деформацией скелета и отставанием в росте. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу.
МПС IVА, ген GALNS локализован в хромосомной области 16q24.3.
МПС IVВ, ген GBS локализован в хромосомной области 3q21.33. Важно отметить, что мутация гена, кодирующего β- галактозидазу, вызывает также ганглиозидоз типа I.
Тип наследования: Наследуется по аутосомно-рецессивному типу.
Распространенность МПС IVА 1:250 000 новорожденных, МПС IVВ встречается еще реже.
E 76.2 – Мукополисахаридоз IV типа
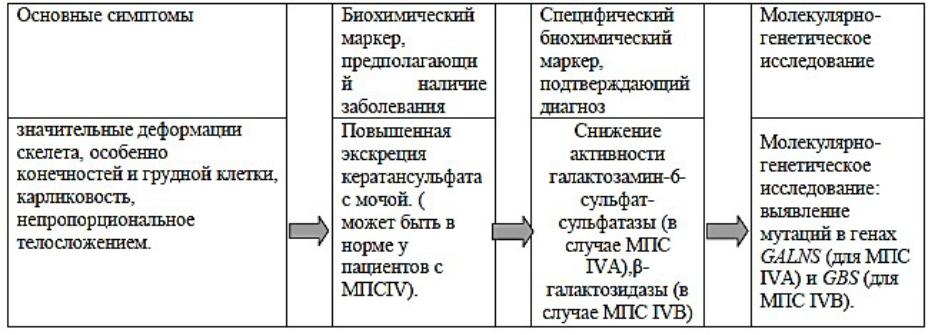
АЛГОРИТМ ДИАГНОСТИКИ МПС IV ТИПА
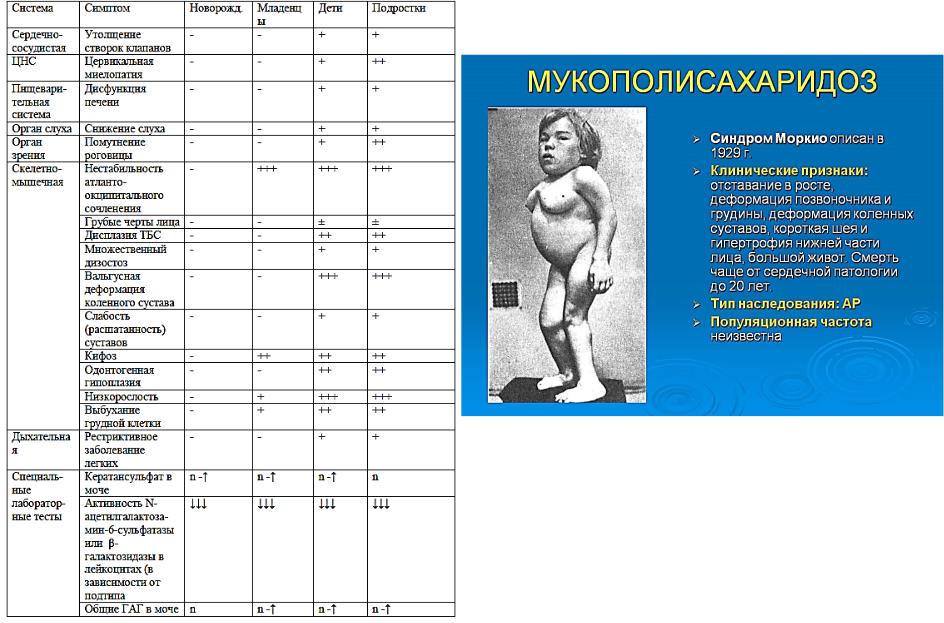
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ В ЗАВИСИМОСТИ ОТ ВОЗРАСТА ДЕБЮТА МПС IV
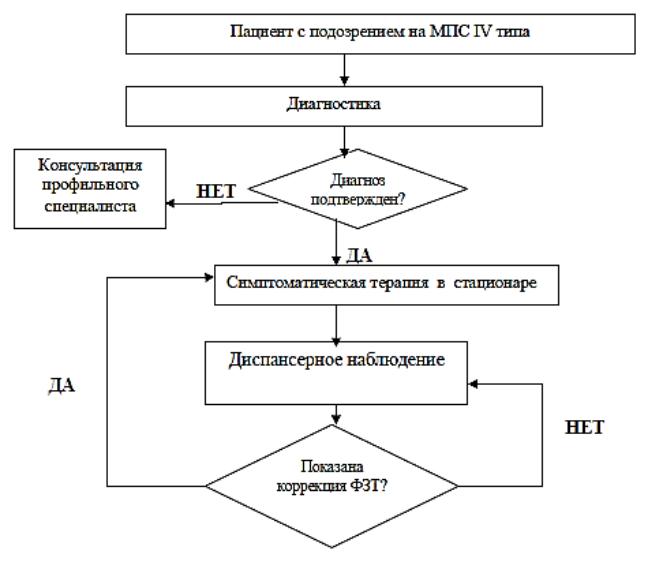
АЛГОРИТМ
ВЕДЕНИЯ
ПАЦИЕНТА
СМПС IV ТИПА
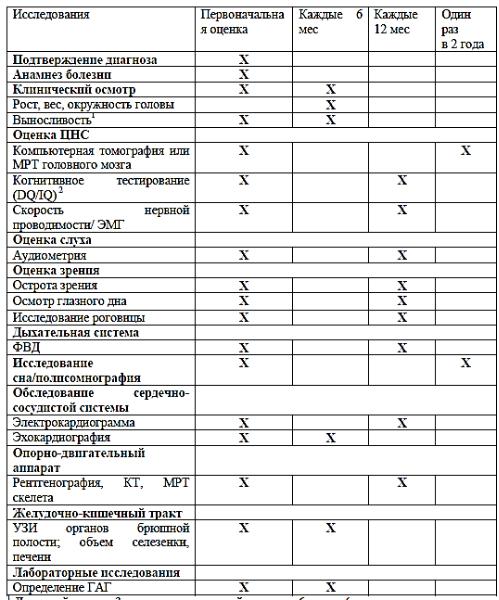
ТАКТИКА ВЕДЕНИЯ ПАЦИЕНТОВ С МПС IV ТИПА
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ МПС IV ТИПА
Ферментозаместительная терапия разработана, однако, в настоящее время лекарственный препарат в Российской Федерации не зарегистрирован. Препарат показал свою эффективность в международных клинических исследованиях. Его назначение возможно по жизненным показаниям на основании решения врачебного консилиума.
Очень важно симптоматическое лечение.
ИСХОДЫ И ПРОГНОЗ
Летальный исход наступает до достижения возраста 20 лет вследствие сердечно-легочной недостаточности на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.

МУКОПОЛИСАХАРИДОЗ VI ТИПА (СИНДРОМ МАРОТО ЛАМИ)
Наследственная лизосомная болезнь накопления, при которой недостаточность фермента N-ацетилгалактозамин-4-сульфатазы (арилсульфатазы В) приводит к нарушению ступенчатой деградации глюкозаминогликана (ГАГ)
дерматансульфата.
Ген ARSB, кодирующий арилсульфатазу В, локализуется в хромосомной области 5q14. Синдром Марото — Лами наследуется по аутосомно- рецессивному типу.
