

5 курс / Детская хирургия / Учебники / Национальное руководство Детская хирургия Разумовский
.pdfЗаболевания толстой кишки 393
Рекомендации по ведению больных ювенильным полипозом приведены в табл. 24.4.
Таблица 24.4. Протокол лечения больных ювенильным полипозом
Колоноскопические полипэктомии каждые 1–3 года
Колэктомия в случае:
•большого количества полипов (не поддающегося консервативному контролю);
•диспластических полипов;
•неконтролируемого кровотечения;
•достижения 20-летнего возраста у большинства пациентов
Рекомендуется выполнение тотальной проктоколэктомии с созданием J-резервуара, учитывая высокую вероятность продолженного роста полипов в сохраненных отделах прямой кишки
Рекомендуется эндоскопическое исследование верхних отделов ЖКТ
Генетический скрининг
Синдромы, при которых обнаруживаются множественные ювенильные полипы
Синдром наследственного смешанного полипоза
Синдром наследственного смешанного полипоза характеризуется атипическими ЮП, аденомами и раком толстой кишки. Thomas и соавт. (1996) указывают на тот факт, что при данном синдроме высок риск вероятности обнаружения воспалительных и метапластических полипов. Наследование происходит по ауто- сомно-доминантному типу. Не вполне ясно, является ли синдром наследственного смешанного полипоза вариантом ювенильного полипоза или отдельным заболеванием. Предположительный генетический локус — 6q.
Синдром Коудена
Синдром Коудена — синдром с аутосомно-доминантным типом наследования, характеризующийся множественными гамартомами, возникающими в любых органах. Классическим примером таких гамартом является трихолеммома. Пациенты с синдромом Коудена имеют высокий риск развития рака молочной и щитовидной (исключая медуллярный рак) желез. Клинические проявления синдрома включают поражения слизистых оболочек и кожи, пороки ЩЖ, фиброзно-кистозную мастопатию, гамартомы ЖКТ, раннее образование лейомиом матки, макроцефалию, задержку умственного развития и диспластическую ганглиоцитому мозжечка (болезнь Лермитта–Дюкло). Синонимы: болезнь Коудена, синдром множественных гамартом. В основе синдрома лежит наследственная мутация гена PTEN/MMAC1.
Синдром был первоначально описан в 1963 г. K.M. Lloyd и M. Dennis в семье Rachel Cowden. P. Weary и соавт. (1972) дали более подробное описание клинических проявлений синдрома и предложили название «синдром множественных гамартом».
Синдром Коудена обычно клинически проявляется к концу второго десятилетия жизни. Синдром имеет различную степень экспрессии и пенетрантности, связанную с возрастом. К третьей декаде жизни у 99% пациентов с синдромом Коудена обнаруживаются кожно-слизистые поражения, хотя другие признаки синдрома уже могут иметь место к этому времени. Предположительно полипы могут обнаруживаться на всем протяжении ЖКТ. В толстой кишке их размеры варьируют от 3 до 10 мм, но могут достигать и 2 см в диаметре. Часть полипов могут представлять простые выпячивания слизистой оболочки, другие могут иметь определенные структурные особенности. Большинство полипов характеризуются наличием участков разрастания соединительной ткани, гладкомышечных клеток, иногда липоцитов. Железы имеют обычное строение, но распределены неравномерно и
24 Глава

Раздел IV
394 |
Абдоминальная хирургия |
часто имеют удлиненную, вытянутую форму. Обнаружение полипов с преобладанием пролиферации нервных клеток и нервных волокон, то есть имеющих ганглионейромоподобное строение, не характерно для синдрома Коудена и описано в виде исключительных случаев. Большинство гамартомных полипов при синдроме Коудена асимптоматичны. Злокачественные опухоли ЖКТ нечасто встречаются у пациентов с синдромом Коудена, однако, по мнению отдельных авторов, у них повышена вероятность развития рака толстой кишки.
Синдром Банаяна–Райли–Рувалькаба
Ранее считался отдельным синдромом, имеющим свои клинические особенности, которые включают макроцефалию, липоматоз, гемангиоматоз и пигментацию полового члена.
В настоящее время входит в состав синдрома Коудена, являясь его аллельным вариантом. Приблизительно 60% семей с синдромом Банаяна–Райли–Рувалькаба
иизолированных случаев синдрома имеют наследственную мутацию гена PTEN. Всего в литературе было описано 11 семей, в которых имело место сочетание клинических проявлений синдрома Коудена и синдрома Банаяна–Райли–Рувалькаба,
ив 10 из них была обнаружена мутация гена PTEN. На основании общности клинических проявлений, высокой вероятности развития злокачественных новообразований и наличия одного и того же генетического дефекта было предложено обозначать данные синдромы как PTEN-ассоциированные синдромы гамартомных опухолей.
Аденоматозные полипы
Если около 70% полипов у взрослых являются аденомами, то у детей солитарные аденоматозные полипы обнаруживаются крайне редко. Обнаружение даже одиночного аденоматозного полипа у ребенка должно насторожить педиатра в отношении первых проявлений синдрома аденоматозного полипоза с высоким риском раннего развития рака толстой кишки и внекишечных поражений.
Семейный аденоматозный полипоз
Семейный аденоматозный полипоз, включая синдром Гарднера, — синдром с аутосомно-доминантным типом наследования, характеризующийся множественными аденоматозными полипами толстой кишки, которые имеют высокий злокачественный потенциал. Синдром Гарднера является вариантом семейного аденоматозного полипоза, к дополнительным проявлениям которого относятся эпидермальные кисты, остеомы, аномалии зубов и десмоиды.
Синонимы: аденоматозный полипоз толстой кишки, семейный полипоз толстой кишки, полипоз аденоматозный кишечный, полипоз Бюссэ–Гарднера.
По разным данным, заболеваемость семейным аденоматозным полипозом варьирует от одного на 7000–30 000 новорожденных.
На сегодняшний день остаются принятыми следующие диагностические критерии синдрома семейного аденоматозного полипоза:
1)100 аденом толстой кишки или более;
2)или наследственная мутация гена АРС;
3)или семейный анамнез синдрома и как минимум один из следующих дополнительных признаков: эпидермоидные кисты, остеомы, десмоиды.
Аденомы в толстой кишке у пациентов с семейным аденоматозным полипозом начинают определяться при проведении эндоскопических исследований в возрасте 10–20 лет (рис. 24.9).
На ранних стадиях формирования аденом течение синдрома может быть бессимптомным. К классическим проявлениям синдрома относятся кишечное кровотечение, диарея со значительной примесью слизи в стуле и боли в живо-
Заболевания толстой кишки 395
24 Глава
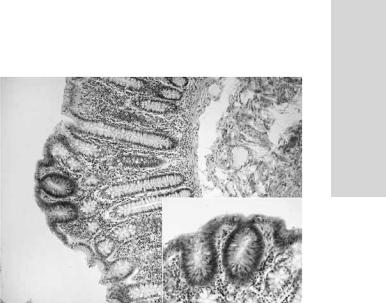
Рис. 24.9. Типичные ранние бикриптальные поражения слизистой оболочки толстой кишки при семейном аденоматозном полипозе
те — типичные проявления всех синдромов полипоза ЖКТ. Симптомы появляются постепенно и легко могут быть просмотрены. Средний возраст развернутых клинических проявлений синдрома у больных, у которых не проводился профилактический скрининг, — 33 года. Следует различать пациентов с семейным аденоматозным полипозом, у которых диагноз ставят на основании клинических проявлений, и тех, кого он был подтвержден в результате скрининга членов семей, страдающих полипозом. В первом случае средний возраст к моменту постановки диагноза составил 36 лет (от 4 до 72 лет), во второй группе — 24 года (от 9 до 57 лет). Средний возраст, когда у больных был выявлен рак толстой кишки, составил 39 лет. Однако риск развития рака уже к возрасту 20–25 лет составляет 1–6%.
Для скрининга в первую очередь используется сигмоскопия с использованием гибкого эндоскопа, которая назначается всем детям от родителей с семейным аденоматозным полипозом в возрасте 10–15 лет и проводится каждые последующие 1–2 года до достижения ими 40-летнего возраста в том случае, если аденомы в течение этого времени не были обнаружены.
Эндоскопия (колоноскопия) — основной метод диагностики и контроля при полипозе. Картина при диффузном полипозе очень характерная. Диагноз САП может быть поставлен, когда у пациента 100 полипов толстой кишки и более. В начальных стадиях заболевания и при ослабленном САП количество полипов может быть меньше. Полипы обычно появляются в 2–3 декаде жизни, от 8 до 34 лет, в среднем в 15 лет. При этом на слизистой оболочке толстой кишки вплотную друг к другу на широком основании располагаются небольшие, округлые полипы розового цвета. Обычно размеры полипов — 3–5 мм, иногда на фоне мелких полипов встречаются более крупные полипы — до 1 см в диаметре, некоторые на ножке (рис. 24.10).
Со временем некоторые из аденом трансформируются в ворсинчатые полипы, поверхность становится яркоили темно-красной, дольчатой. В 70–80% таких полипов обнаруживается малигнизация. При отсутствии лечения к 39 годам может развиться аденокарцинома, к 45 годам у 89% пациентов развивается рак и к 50 годам у 93% пациентов наблюдается аденокарцинома. Если полипов в толстой кишке более 1000, то риск развития карциномы у таких пациентов в 2 раза выше.
Проводится исследование обязательно всей толстой кишки, с биопсией полипов в каждом отделе кишки, при этом оценивается степень пораженности различных ее отделов. В редких случаях, при невозможности осмотреть всю толстую кишку,

Раздел IV
396 |
Абдоминальная хирургия |
|
|
|
|
проводится исследование толстой кишки |
|
|
|
с двойным контрастированием бариевой |
|
|
|
взвесью и воздухом. |
|
|
|
Полипы также развиваются в других |
|
|
|
отделах ЖКТ. Полипы в желудке встреча- |
|
|
|
ются у 23–100% пациентов, обычно это |
|
|
|
ненеопластические железистые полипы |
|
|
|
(рис. 24.11). Полипы могут покрывать всю |
|
|
|
слизистую оболочку дна и тела желудка. |
|
Рис. 24.10. Семейный аденоматозный поли- |
На поверхности полипов может выявлять- |
||
ся дисплазия, которая в редких случаях |
|||
поз. Нисходящий отдел толстой кишки |
малигнизируется. Существует повышенный |
||
|
|
риск развития рака желудка, однако в тече- |
|
|
|
ние жизни этот риск уменьшается менее |
|
|
|
чем до 1%. |
|
|
|
Полипы ДПК, включающие ампулярные |
|
|
|
полипы, клинически более значимы, чем |
|
|
|
полипы желудка. У 50–90% пациентов с |
|
|
|
САП развиваются полипы в ДПК, размером |
|
|
|
1–5 мм, единичные, по всей кишке, чаще в |
|
|
|
преампулярном отделе (рис. 24.12). Как и в |
|
|
|
толстой кишке, полипы ДПК демонстриру- |
|
|
|
ют карциноматозную прогрессию и могут |
|
Рис. 24.11. Железистые полипы дна желуд- |
иметь ворсинчатую структуру с высокой |
||
степенью дисплазии. Дуоденальный рак |
|||
ка у пациента с САП |
|||
|
|
развивается в 3–5% случаев, чаще в воз- |
|
|
|
расте 45–52 лет. Это наиболее частая при- |
|
|
|
чина смерти у пациентов, подвергшихся |
|
|
|
колэктомии. |
|
|
|
Аденомы в тонкой кишке встречаются у |
|
|
|
50% пациентов, в основном в проксималь- |
|
|
|
ном отделе тощей кишки или в дистальном |
|
|
|
отделе подвздошной (20%). Также после |
|
|
|
колэктомии полипы могут развиваться в |
|
|
|
дистальном отделе подвздошной кишки. |
|
|
|
Рак тонкой кишки встречается редко. |
|
|
|
Крайне редко полипы могут развиваться в |
|
Рис. |
24.12. Полипы двенадцатиперстной |
желчном пузыре, желчных протоках, ПЖ. |
|
Эндоскопия может быть заменена гене- |
|||
кишки у пациента с САП |
тическим тестированием для обнаружения |
||
|
|
||
специфической мутации гена АРС, которая имеет место в данной семье. В этом случае позитивный тест является диагностическим.
Американской гастроэнтерологической ассоциацией были разработаны рекомендации по проведению скрининга и по тактике лечения больных семейным аденоматозным полипозом.
Рекомендации по проведению скрининга:
•• поиск мутаций у ближайших родственников больного в возрасте 10 лет — чувствительность метода составляет 80–90%;
•• при отсутствии мутации — проведение периодических эндоскопических исследований с использованием гибкого сигмоскопа начиная с 12-летнего

Заболевания толстой кишки 397
возраста и до 50 лет, затем — рутинные исследования по выявлению рака толстой кишки;
•• эзофагогастродуоденоскопия каждые 6 мес — 4 года и ежегодное обследование ЩЖ;
•• определение уровня α-фетопротеина в сыворотке крови у детей с семейным анамнезом, учитывая высокий риск (1:150) развития гепатобластомы;
•• молекулярные методы диагностики и их чувствительность:
анализ сцепленного наследования (требует наличия двух больных членов одной семьи) — чувствительность 99%;
тест по определению структурных особенностей АРС-белка (Protein Truncation Test — PTT) — 80–90%;
конверсионный тест (conversion PTT) — 96%;секвенирование генома (sequencing) — 90%.
Рекомендации по лечению:
•• колэктомия после постановки диагноза, ее проведение может быть отложено у подростков с аденомами, не превышающими 0,6 мм в диаметре;
•• назначение ингибиторов циклооксигеназы (COX-2 inhibitors);
•• сохранение прямой кишки и предупреждение или элиминация аденом дают только кратковременный эффект;
•• создание резервуара из подвздошной кишки — на сегодняшний день данных недостаточно для оценки эффективности данного вида оперативного вмешательства;
•• аденомы ДПК — подходы не разработаны (в небольших исследованиях не было получено обнадеживающих результатов).
При отсутствии полипов в прямой кишке операцией выбора является колэктомия с частичной резекцией прямой кишки и илеоректальным анастомозом. При наличии полипов сразу выше ануса показана колэктомия с демукозацией прямой кишки и илеоанальным анастомозом.
В настоящее время развитие эндохирургии позволяет проводить довольно сложные операции минимально травматично, что значительно облегчает послеоперационный период, сокращает сроки госпитализации и реабилитации. Основной принцип лечения полипозов в детском возрасте — сохранение замыкательного аппарата прямой кишки.
При демукозации прямой кишки оперативное лечение заканчивается илеоректальным анастомозом, который можно сформировать одномоментно либо вывести тонкую кишку через серозно-мышечный цилиндр прямой кишки на промежность, фиксировать ее за серозную оболочку узловыми швами к терминальной части слизистой прямой кишки и оставить культю свободно висящей. Культю отсекают на 14–15-е сутки, как при операции Соавэ–Ленюшкина.
Некоторые авторы рекомендуют формировать тонкокишечные J- или S-резер вуары, что усложняет оперативное лечение, повышает риск развития осложнений, требует формирования петлевой илеостомы, осложняет послеоперационный эндоскопический контроль за низведенной кишкой и анастомозом.
Через несколько месяцев после наложения прямого илеоректального анастомоза обычно кишечник адаптируется к новым условиям, стул становится менее частым и разжиженным, опорожнение происходит 3–4 раза в сутки; благодаря сохраненному замыкательному аппарату со временем формируется ампула прямой кишки, ребенок удерживает каловые массы, может свободно посещать школу, общаться со сверстниками.
24 Глава

Раздел IV
398 |
Абдоминальная хирургия |
Синдром Гарднера
В начале 50-х гг. ХХ в. генетик Eldon J. Gardner и соавт. сообщили о группе пациентов со множественными аденомами толстой кишки и семейным раком толстой кишки, как и при семейном аденоматозном полипозе, но также имевших остеомы костей черепа и нижней челюсти, множественные эпидермальные кисты и другие поражения кожи. Позже к проявлениям синдрома были добавлены аномалии дентина, одонтогенные кисты, аномальная структура кости нижней челюсти и десмоидные опухоли.
Существует по меньшей мере три аргумента против разделения синдрома Гарднера и семейного аденоматозного полипоза: 1) ошибочная тенденция считать любые внетолстокишечные проявления семейного аденоматозного полипоза как проявления синдрома Гарднера; 2) тщательный семейный анамнез, который показал наличие в семьях больных семейным аденоматозным полипозом родственников с проявлениями синдрома Гарднера и отсутствие у родственников больных синдромом Гарднера каких-либо внекишечных проявлений; 3) сообщения, в которых у больных семейным аденоматозным полипозом при тщательном обследовании были обнаружены признаки синдрома Гарднера.
Синдром Тюрко
Синдром Тюрко — синдром с аутосомно-доминантным типом наследования, характеризующийся развитием полипов или рака толстой кишки в сочетании со злокачественными нейроэпителиальными опухолями, обычно медуллобластомами и глиобластомами. Большинство случаев синдрома Тюрко входят в состав семейного аденоматозного полипоза или наследственного, не связанного с полипозом рака толстой кишки.
На связь нейроэпителиальных опухолей и полипов толстой кишки впервые обратил внимание H.W. Crail. Было предложено выделять три группы больных при сочетании поражений толстой кишки и ЦНС: 1) тех, у которых братья или сестры (двое или более) имеют множественные полипы толстой кишки и злокачественные опухоли мозга, но ни родители, ни другие родственники не имеют клинических проявлений синдрома; 2) больных, имеющих аутосомно-доминантный синдром наследственного полипоза с поражением нескольких поколений в семье; 3) изолированные несемейные случаи. Эти данные позволили предположить, что синдром Тюрко может являться вариантом семейного аденоматозного полипоза.
Синдром Тюрко является гетерогенным и состоит из как минимум двух синдромов. Синдром Тюрко 1-го типа включает глиобластому у больных без семейного аденоматозного полипоза, часть из которых имеют наследственный, не связанный с полипозом рак толстой кишки и наследственные мутации генов hPMS2, hMSH2 или MHL1. Синдром Тюрко 2-го типа характеризуется развитием медуллобластомы у больных семейным аденоматозным полипозом и наследственной мутацией гена АРС. Начиная с 1949 г. в литературе было опубликовано приблизительно 160 случаев синдрома.
Клинические проявления синдрома со стороны ЖКТ включают два основных варианта. Синдром Тюрко 1-го типа характеризуется небольшим количеством крупных полипов толстой кишки и развитием рака толстой кишки в молодом возрасте в 56% случаев. Синдром Тюрко 2-го типа характеризуется множественными аденомами толстой кишки и относительно низкой долей (21%) развития рака толстой кишки. Поражения кожи описаны у 50% пациентов с 1-м типом синдрома и у 20% пациентов с 2-м типом. Пятна цвета кофе с молоком были обнаружены у 38% пациентов с синдромом 1-го типа. Костно-лицевые экзостозы и врожденная гипертрофия пигментного эпителия слизистой оболочки толстой кишки наблюда-



