

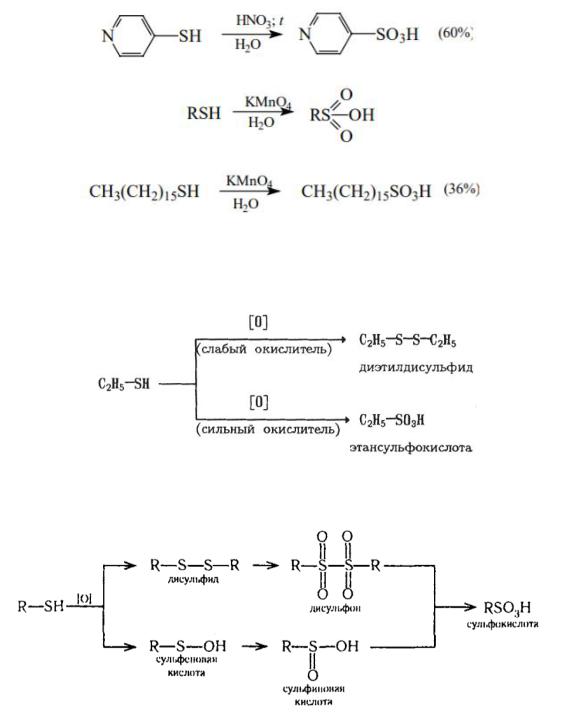
483
Сильные окислители – азотная кислота или перманганат калия окисляют тиолы до сульфоновых кислот (продуктов исчерпывающего
окисления органических соединений серы):
Образование дисульфидов и их роль в биохимических процессах
Действие слабых окислителей на меркаптаны при водит к образованию дисульфидов. Сильные окислители дают сульфокислоты:
Таким образом, в отличие от спиртов при окислении тиолов окисляется не углерод, а сера. Реакция идет, вероятно, по одному из следующих путей:
У бактерий и архей
Дисульфидные связи играют важную защитную роль для бактерий в качестве обратимого переключателя, который включает или выключает белок, когда бактериальные клетки подвергаются реакциям окисления. В частности, перекись водорода (H2O2) могла серьезно повредить ДНК и убить бактерии при низких концентрациях, если бы не защитное действие S-S- связи. Археи обычно содержат меньше дисульфидов, чем высшие организмы.
У эукариот
484
В эукариотических клетках, как правило, стабильные дисульфидные связи образуются в просвете RER (грубый эндоплазматический ретикулум) и митохондриальном межмембранном пространстве, но не в цитозоле. Дисульфидные связи в основном обнаруживаются в секреторных белках, лизосомальных белках и экзоплазматических доменах мембранных белков. Из этого правила есть заметные исключения. Например, многие ядерные и цитозольные белки могут стать сшитыми дисульфидом во время некротической гибели клеток. Точно так же ряд цитозольных белков, которые имеют близкие друг к другу остатки цистеина, которые действуют как сенсоры окисления или окислительно-восстановительные катализаторы; когда восстановительный потенциал клетки не работает, они окисляются и запускают механизмы клеточного ответа. Вирус осповакцины также продуцирует цитозольные белки и пептиды, содержащие множество дисульфидных связей; хотя причина этого неизвестна, по-видимому, они обладают защитным действием против аппарата внутриклеточного протеолиза. Дисульфидные связи также образуются внутри и между протаминами в хроматине сперматозоидов многих видов млекопитающих.
3.6.4 Амины
Классификация
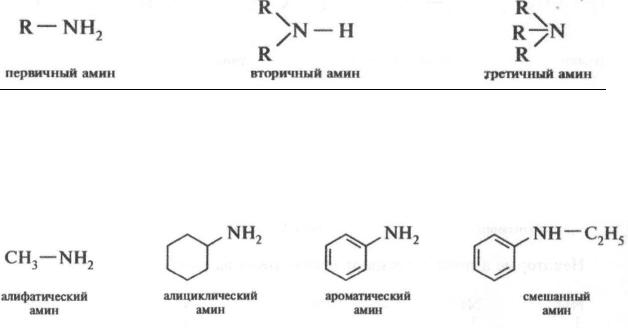
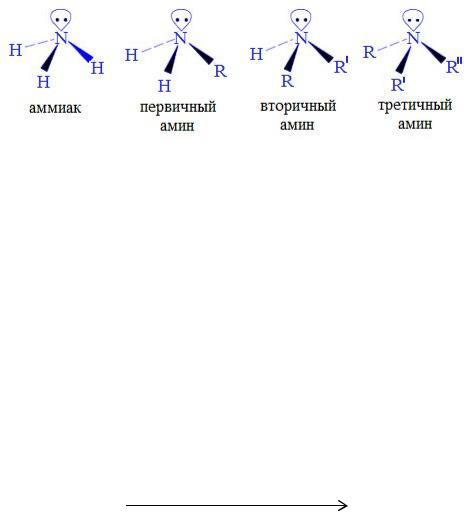
Аминами называют производные аммиака, в молекуле которого один, два или три атома водорода замещены углеводородными радикалами.
Соответственно числу углеводородных остатков различают первичные,
вторичные и третичные амины.
В зависимости от природы углеводородных радикалов у атома азота амины подразделяют на алифатические, алициклические и ароматические
(аренамины); амины, у которых атом азота связан с алифатическим и ароматическим углеводородным радикалом, называют смешанными:
Номенклатура
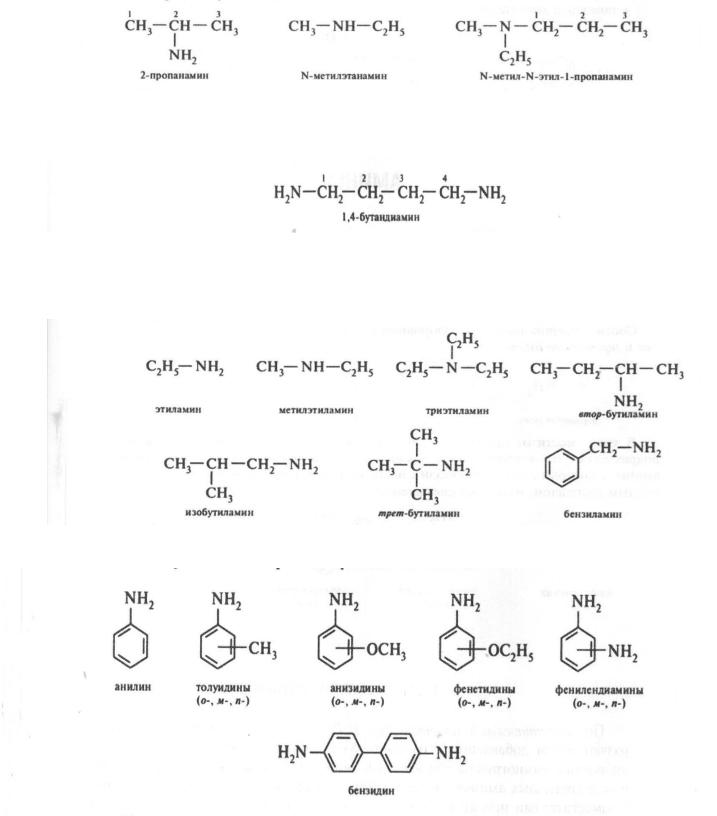
По заместительной номенклатуре IUPAC названия первичных аминов образуют путем добавления к названию углеводорода суффикса -амин, указывая положение аминогруппы в углеродной цепи. При составлении
485
названий вторичных и третичных аминов их рассматривают как производные первичного амина с заместителями при атоме азота. За исходный первичный амин в этом случае принимается связанный с атомом азота наиболее сложный по структуре радикал. Остальные углеводородные заместители при атоме азота перечисляют в алфавитном порядке с указанием локанта N-:
Если соединение содержит две или три аминогруппы, то в названии их обозначают множительными приставками ди- или три-, которые ставятся перед суффиксом -амин:
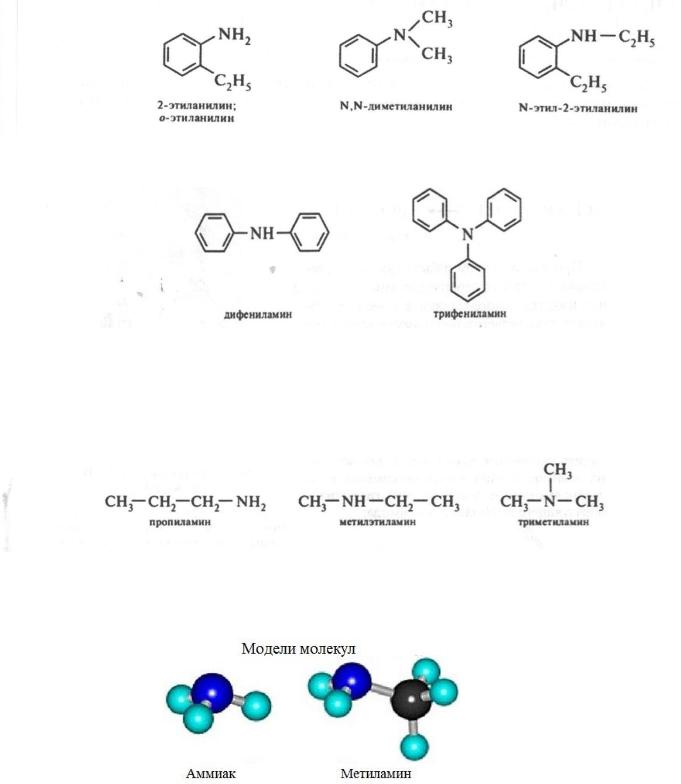
Простейшие амины чаще всего называют по радикало-функциональной номенклатуре. Согласно этой номенклатуре, названия аминов образуют из названий углеводородных радикалов, перечисляемых в алфавитном порядке, и суффикса -амин:
Некоторые амины сохраняют тривиальные названия:
487
пространстве). Три из четырех гибридных орбиталей участвуют в образовании σ-связей N-C и N-H.
На четвертой орбитали находится неподеленная электронная пара, которая обусловливает основные свойства аминов:
Два неспаренных электрона в молекулах аминов могут участвовать в образовании химической связи по донорно-акцепторному механизму.
У первичных аминов метильная группа при связи углерода с более электроотрицательным атомом проявляет положительный индуктивный эффект (+I) и, следовательно, смещает от себя электронную плотность в сторону атома азота, повышая тем самым электронную плотность неподеленной электронной пары.
По этой причине электронная пара атома азота удерживается менее прочно и легче взаимодействует с протоном. Она становится доступной для атаки реагентов, имеющих вакантную орбиталь и облегчает образование донорно-акцепторной связи с неподеленной электронной парой азота.
Следовательно, первичные амины более сильные основания, чем аммиак.
NH3 |
CH3-NH2 |
аммиак |
первичные амины |
Увеличение основных свойств
Их основные свойства усиливаются при удлинении углеводородного радикала.
У вторичных аминов две алкильные группы будут давать двойной положительный индуктивный эффект, еще больше концентрируя электронную плотность на неподеленной электронной паре азота.
Основные свойства у вторичных аминов будут выражены сильнее, чем
упервичных аминов.
Втретичных аминах важную роль играет стерический (пространственный) фактор. Хотя электронная плотность на азоте попрежнему высока, но три объемных заместителя загораживают электронную пару атома азота и затрудняют ее взаимодействие с другими молекулами. Присоединение протона происходит не так эффективно.
По этой же причине основность первичных и вторичных аминов снижается с увеличением размеров и разветвленности радикалов.
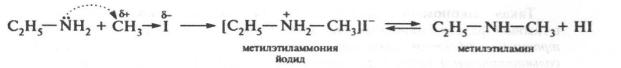
488
Третичные амины являются более слабыми основаниями, чем первичные и вторичные амины. Но более сильными, чем аммиак.
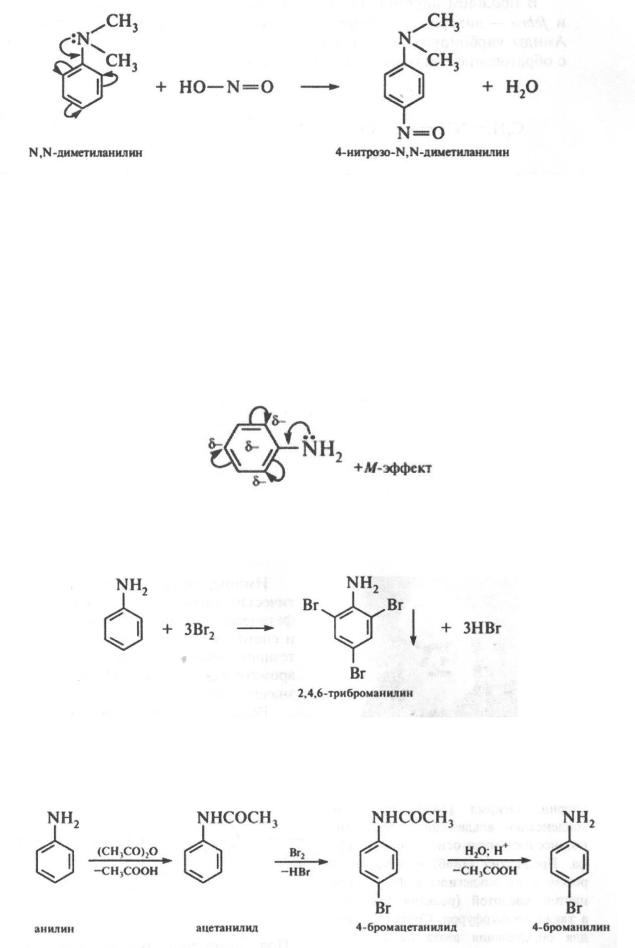
Ароматические амины более слабые основания, чем аммиак, так как электронная пара азота втягивается в бензольное кольцо, вследствие чего снижается способность неподеленной пары электронов азота присоединять протон. Поэтому анилин как основание слабее, чем аммиак, а дифениламин слабее, чем анилин.
Электронодонорные заместители (алкил, -ОСН3, -N (СН3)2 и др.) увеличивают основность аминов, а электроноакцепторные заместители (-F, -Cl, -NO2 и т.п.) – уменьшают.
Реакции ацилирования и алкилирования аминов. Аммониевые
соли.
Алкиламинами называют продукты замещения одного, двух или трех атомов водорода в аммиаке алкильными группами.
Взаимодействие с галогеналканами. С помощью этой реакции в структуру амина вводят алкильный заместитель, и поэтому она получила название «реакции алкилирования». При взаимодействии с галогеналканами первичные амины превращаются во вторичные, вторичные –в третичные, а третичные –образуют четвертичные аммониевые соли:
Аналогично вторичный амин алкилируется с образованием третичного, а третичный – с образованием четвертичной аммониевой соли.
В зависимости от природы галогеналкана и алкиламина реакция может протекать по механизму SN1 или SN2.
Ацилирование. Первичные и вторичные алкиламины вступают в реакцию с функциональными производными карбоновых кислот – галогенангидридами, ангидридами или сложными эфирами, образуя соответствующие амиды:

489
В процессе реакции атом водорода при атоме азота в молекуле амина замещается на остаток карбоновой кислоты, называемый ацильной группой.
Реакции, с помощью которых в молекулу органического вещества вводится ацильная группа, называют реакциями ацилирования.
Третичные амины не содержат при атоме азота атома водорода и поэтому в реакцию ацилирования не вступают.
Взаимодействие с азотистой кислотой. С азотистой кислотой реагируют первичные и вторичные алкиламины.
При действии азотистой кислоты на первичные алкиламины выделяется свободный азот и образуются спирты:
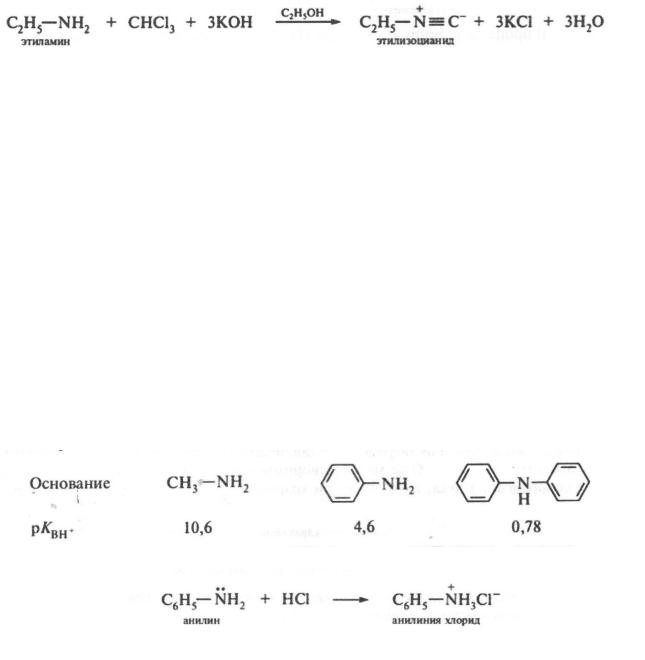
Вторичные алкиламины в реакции с азотистой кислотой образуют N- нитрозамины:
Нитрозамины представляют собой желтые или оранжевые маслянистые жидкости. При обработке концентрированными минеральными кислотами они расщепляются с образованием исходного амина и азотистой кислоты:
Тр етичные алкиламины в обычных условиях с азотистой кислотой не реагируют.
Реакция взаимодействия алкиламинов с азотистой кислотой может
492
Реакции с участием ароматического ядра
Для ариламинов характерны реакции электрофильного замещения по ароматическому ядру, свойственные ароматическим углеводородам. Аминогруппа в молекуле ариламина в результате проявления +M-эффекта выступает в качестве сильного электронодонорного заместителя по отношению к бензольному кольцу и тем самым активизирует его реакционную способность в реакциях электрофильного замещения. Поэтому ариламины вступают в реакции электрофильного замещения значительно легче, чем бензол. Аминогруппа, являясь заместителем I-рода, направляет электрофильное замещение в орто- и пара-положения.
Галогеиирование. Анилин легко реагирует с галогенами (С12, Вг2) в отсутствие катализатора, образуя 2,4,6-тригалогенопроизводные ариламины.
При получении моногалогенозамещенных ариламинов амины первоначально переводят в N-ацильные производные, которые затем галогенируют и гидролизуют:
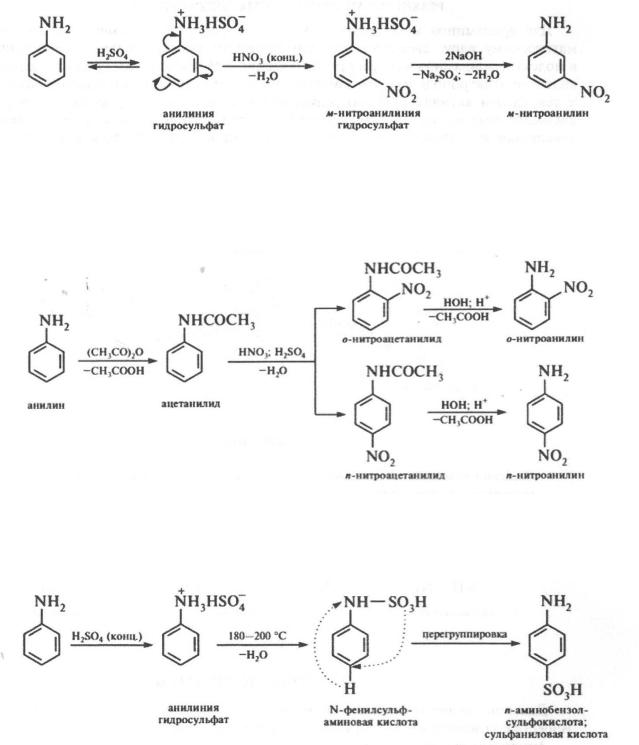
493
Нитрование. Нитрование ариламинов в отличие от аренов имеет ряд особенностей. Прямое нитрование ароматических аминов концентрированной азотной кислотой осуществить невозможно, так как они легко окисляются. При использовании в качестве нитрующего реагента нитрующей смеси ариламины, наряду с частично протекающими окислительными процессами, превращаются в ариламмонийные соли. Аммонийная группа, являясь электроноакцепторным заместителем, затрудняет нитрование и способствует образованию преимущественно мета- изомера:
С целью защиты аминогруппы от процессов окисления и протонирования по атому азота ароматические амины предварительно ацетилируют. Соотношение орто- и пара-изомеров зависит от состава нитрующей смеси и условий проведения реакции. После нитрования N- ацильные производные гидролизуют в кислой или щелочной среде:
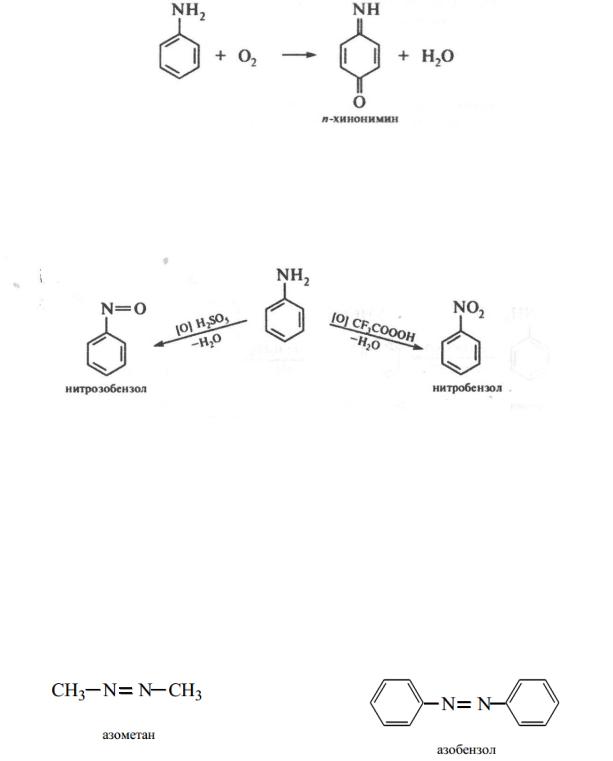
Сульфирование. При нагревании анилина с концентрированной серной кислотой в среде высококипящего растворителя образуется n- аминобензолсульфокислота, которую чаще называют сульфаниловой кислотой:
495
2-гидрокси-3`-метилазобензол, о-гидрокси-м`-метилазобензол
Диазосоединения
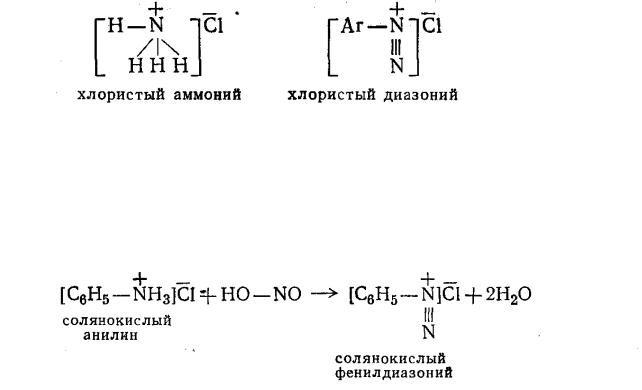
По своему строению диазосоединения ArN2X делятся на две группы:
1)собственно диазосоединения, в которых оба атома азота трехвалентны, имеют строение Аr — N = N — Х (где Ar— остаток ароматического углеводорода, или арил), а Х — кислотный остаток, гидроксил, или радикал);
2)соли диазония, в которых один атом азота трехвалентен, а другой, у как в аммонийной группе, имеет 4 ковалентных связи и входит в состав иона, несущего положительный заряд.
Номенклатура диазосоединений Ar—N=N—X :
Углеводород + -диазо + Х:
Номенклатура солей диазония:
окончание –диазоний с указанием аниона (хлорид, сульфат….) :
Собственно, диазосоединения могут существовать в виде двух стереоизомерных форм, называемых син-диазосоединениями и анти- диазосоединениями:
496
Син-диазосоединения очень непрочны, они очень легко вступают в различные соединения и, в частности, легко перегруппировываются в анти- диазосоединения, обладающие гораздо большей прочностью и меньшей реакционной способностью. Собственно диазосоединения могут быть получены из солей диазония.
Наибольшее значение из диазосоединений имеют так называемые соли диазония, по своему строению аналогичные солям аммония. Соли диазония можно рассматривать как производные солей аммония, у которых один атом водорода замещен остатком ароматического углеводо-рода, а остальные три атома водорода – одним атомом трехвалентного азота.
Сходство солей аммония и солей диазония хорошо видно из сопоставления их формул строения:
Таким образом, в солях диазония один атом азота трехвалентен, а другой, связанный четырьмя ковалентными связями с арилом и атомом азота, входит в состав иона диазония, который связан электровалентной связью с анионом кислоты (так же, как в солях аммония или солях аминов).
Соли диазония получаются при действии азотистой кислоты на раствор соли ароматического амина в кислой среде, например:
Соли диазония – очень непрочные соединения и поэтому их обычно не удается выделить из водных растворов. Однако при проведении реакции диазотирования в спиртовом растворе соли диазония можно выделить в твердом кристаллическом состоянии. При легком нагревании или ударе сухие соли диазония (например, солянокислый фенилдиазоний) взрываются с очень большой силой. В связи с этим все реакции с солями диазония обычно проводят с их водными растворами тотчас после образования солей, без выделения их в твердом состоянии.
Соли диазония – вещества, обладающие очень высокой реакционной способностью; через них можно получить целый ряд соединений. Многочисленные реакции солей диазония для удобства рассмотрения обычно делят на две группы: реакции, идущие с выделением азота, и реакции, идущие без выделения азота.
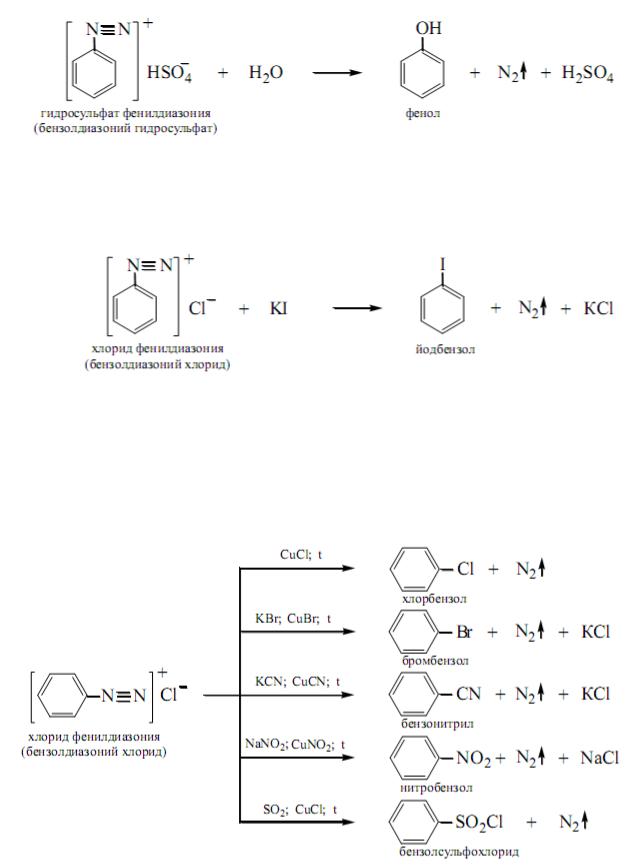
497
I. Реакции, идущие с выделением азота
1. Образование фенолов. При кипячении солей диазония с водой происходит бурное выделение азота и образование фенолов, например:
2. Образование галогенопроизводных (реакция Зандмейера).
При нагревании солей диазония с солями галогеноводородных кислот (KJ, КВг, КС1) происходит выделение азота и замещение диазогруппы —N = N на атом галогена, например:
С достаточным выходом идет образование лишь иодзамещенных. Для получения других галогенозамещенных реакцию проводят в присутствии катализаторов — солей одновалентной меди (CuCl, CuBr) или же мелко раздробленной металлической меди.
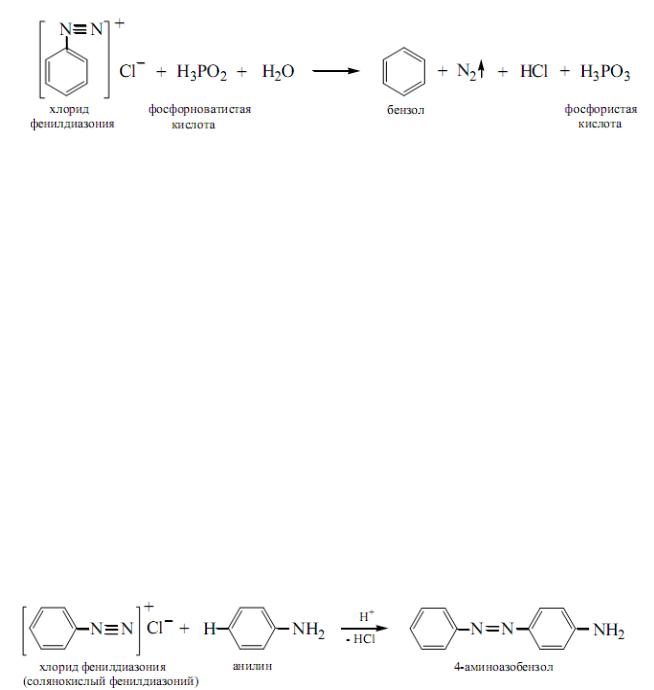
3. Замещение диазогруппы, катализируемое солями одновалентной меди (реакция Зандмейера)
499
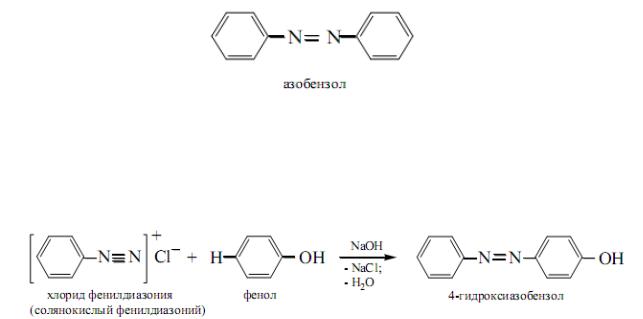
б) Реакция сочетания солей диазония с фенолами. В молекулах фенолов атом водорода, находящийся в пара-положении по отношению к гидроксилу, отличается значительной подвижностью. При взаимодействии солей диазония с фенолами происходит образование азокрасителей подобно тому, как и в реакции с третичными ароматическими аминами:
Краситель, полученный из солянокислого фенилдиазония и фенола, можно рассматривать как азобензол, в котором атом водорода замещен гидроксилом, или оксигруппой, а поэтому его называют оксиазобензолом. Окси-азобензол – азокраситель оранжевого цвета.
Применяя различные первые компоненты красителей (т. е. диазотируемые ароматические амины) и различные вторые компоненты красителей (т. е. различные замещенные аминов и фенолов), можно получить самые разнообразные азокрасители, обладающие различными цветами и оттенками. В настоящее время известны многие сотни различных азокрасителей, обладающих всеми цветами спектра; преобладают среди них желтые, оранжевые и красные.
Для повышения растворимости азокрасителей часто в их молекулы вводят сульфогруппы, для чего, например, диазотируют сульфаниловую кислоту, а затем полученную соль диазония сочетают с аминами или фенолами.
Соль диазония в реакции азосочетания называют диазосоставляющей (диазокомпонентой), а фенол или ароматический амин – азосоставляющей (азокомпонентой). Используя в реакции азосочетания различные диазо- и азосоставляющие можно получить большое число азосоединений.
По числу азогрупп в молекуле различают моно-, дис-и полиазокрасители. Цвет моно-азокрасителей определяется химическим строением связанных азогруппой остатков, числом и положением в них заместителей. Наиболее практически важные моно-азокрасители, содержащие один электронодонорный заместитель, имеют следующие цвета: желтый – производные бензолазоацетоацетарилидов, пиразолоназобензола и азобензола, например, пигмент желтый светопрочный (формула I), жирорастворимый желтый 3 (II); оранжевый и красный-производные соотв. нафталиназобензола и азонафталина, например, кислотный оранжевый (III) и кислотный красный 2С (IV).
500
2. Восстановление солей диазония. Эта реакция идет с разрывом тройной связи между атомами азота и приводит к ароматическим гидразинам, например,
Ароматические производные гидразинов широко применяются в химии карбонильных соединений (в частности, сахаров) для получения гидразонов, а также при ряде синтезов в качестве сильных восстановителей. Фенилгидразин – почти бесцветная жидкость, быстро темнеющая на воздухе вследствие окисления. Солянокислая соль фенилгидразина – кристаллическое вещество, плавящееся при 243°С.
3.6.5 Карбонильные соединения
Классификация, номенклатура и изомерия карбонильных соединений
Органические соединения, содержащие в составе своей молекулы полярную карбонильную (оксо-) группу -С=О, называются карбонильными,
или оксосоединениями.
В зависимости от заместителей, связанных с оксогруппой, эти вещества подразделяют на альдегиды и кетоны.
501
Альдегидами называются соединения, в которых карбонильная группа связана с двумя атомами водорода или с одним атомом водорода и одним атомом углерода.
Кетонами называются соединения, в которых карбонильная группа соединена с двумя атомами углерода.
Общая формула предельных карбонильных соединений СnН2nО.
Классификация
По количеству карбонильных групп:
По типу радикалов:
•Предельные
•Непредельные
•Карбоциклические
•Ароматические
•Гетероциклические
•Смешанные
Номенклатура
Для названия оксосоединений используют следующие номенклатуры:
o |
тривиальную; |
o |
рациональную; |
o |
систематическую. |
По тривиальной номенклатуре альдегиды называют по названию соответствующей карбоновой кислоты. Кроме того, альдегиды могут иметь собственные, исторически сложившиеся названия.
Рациональная номенклатура характерна для альдегидов, имеющих число углеродных атомов в скелете больше двух. Такие альдегиды рассматриваются как производные уксусного альдегида, в котором атомы водорода замещены на углеводородные радикалы. Кетоны в рациональной номенклатуре называют по радикалам, связанным с карбонильной группой.

502
В систематической номенклатуре название строится от исходного насыщенного углеводорода. Альдегидная группа получает первый номер, а кетонная группа наименьший. Альдегидная группа в главной цепи обозначается суффиксом «аль», кетонная группа обозначается суффиксом «он». Но если альдегидная или кетонная группа входят в состав бифункционального соединения, в котором есть группа, которая обозначается позже, то обозначение системы карбонильной группы вводится в префикс и обозначается альдегидная группа «формил», кетонная группа «оксо». В иерархии функциональных групп альдегидная группа обозначается после кетонной, а они обе обозначаются после гидрокси-группы, кратной связи, алкила и галоида (таблица 3.6.5.1, таблица 3.6.5.2).
Таблица 3.6.5.1 – Названия альдегидов по различным номенклатурам
Формула |
|
Номенклатура |
|
|
|
|
|
||
тривиальная |
рациональная |
систематическая |
||
|
||||
|
|
|
|
|
НСHO |
муравьиный альдегид |
формальдегид |
метаналь |
|
|
|
|
|
|
CН3СHO |
уксусный альдегид |
ацетальдегид |
этаналь |
|
|
|
|
|
|
|
пропионовый |
метилацетальдегид, |
|
|
CН3CН2СHO |
метилуксусный |
пропаналь |
||
альдегид |
||||
|
альдегид |
|
||
|
|
|
||
|
|
этилацетальдегид, |
|
|
CН3(CН2)2СHO |
масляный альдегид |
этилуксусный |
бутаналь |
|
|
|
альдегид |
|
|
|
|
диметилаце- |
|
|
CН3CН(CН3)СHO |
изомасляный |
тальдегид, |
2-метилпропаналь |
|
альдегид |
диметилуксусный |
|||
|
|
|||
|
|
альдегид |
|
|
|
валериановый |
пропилацетальдегид |
|
|
CН3(CН2)3СHO |
, пропилуксусный |
пентаналь |
||
альдегид |
||||
|
альдегид |
|
||
|
|
|
||
|
|
изопропилаце- |
|
|
CН3CН(CН3)CН2СH |
изовалериановый |
тальдегид, |
3-метилбутаналь |
|
O |
альдегид |
изопропилуксусный |
||
|
||||
|
|
альдегид |
|
|
|
|
триметилаце- |
|
|
C(CН3)3СHO |
пиваловый альдегид |
тальдегид, |
2,2-диметилпропаналь |
|
триметилуксусный |
||||
|
|
|
||
|
|
альдегид |
|
|
C6Н5СHO |
– |
бензойный альдегид |
бензальдегид |
|
|
|
|
|
|
CН2 = CНСHO |
акролеин |
– |
пропен-2-аль |
|
|
|
|
|
|
CН3CН2 = CНСHO |
кротоновый альдегид |
– |
бутен-2-аль |
|
|
|
|
|
|
C6Н5CН = CНСHO |
коричный альдегид |
– |
3-фенилпропаналь |
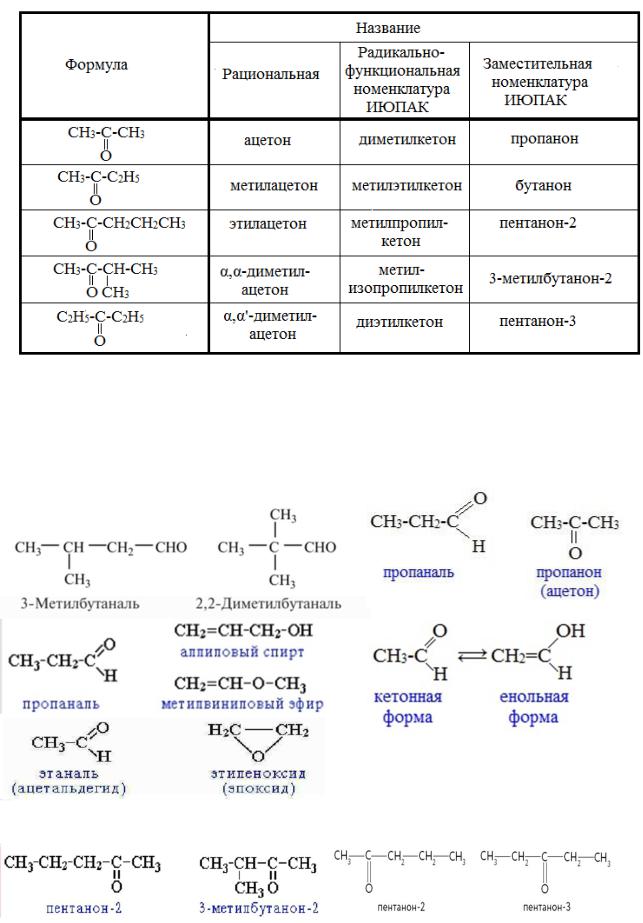
503
Таблица 3.6.5.2 – Названия кетонов по различным номенклатурам
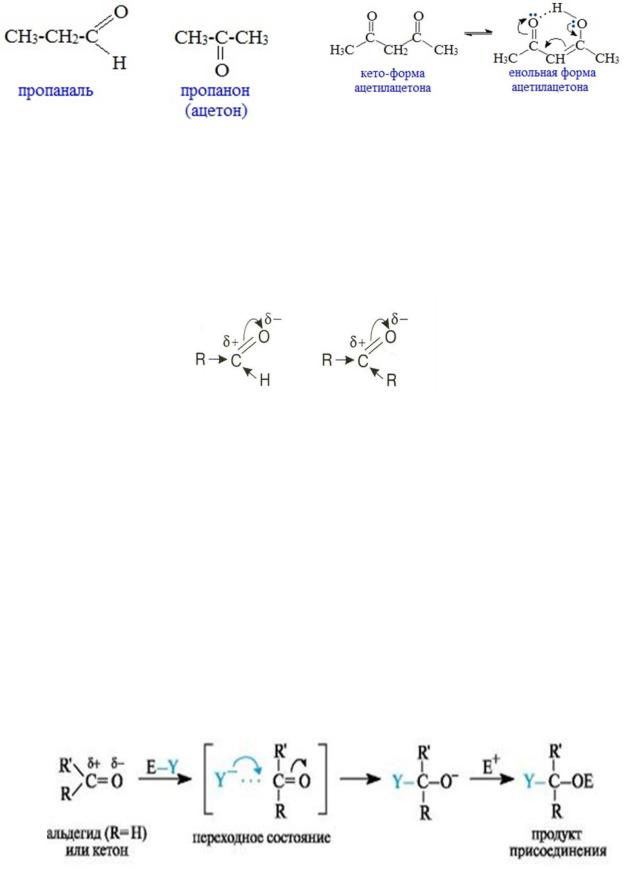
Изомерия
Изомерия для альдегидов и кетонов сводится к изомерии углеродного скелета, начиная с С4, а также межклассовая изомерия начиная с С2, енолизация, кето-енольная таутомерия.
Примеры:
Для кетонов характерна изомерия положения карбонильной группы.
505
гидрирования (Ni, Co, Cu, Pt, Pd и др.), а также литийалюминий гидридом (LiAlH4). При этом альдегиды образуют первичные спирты, а кетоны – вторичные:
|
|
O |
kat |
|
|
|
H C |
C |
+ H2 |
H C |
CH |
|
|
|
OH |
|||||
3 |
|
|
||||
|
|
H |
|
3 |
2 |
|
|
|
|
|
|
|
|
|
O |
kat |
|
OH |
|
H C |
C |
+ H |
H C |
|||
|
CH |
|||||
3 |
|
2 |
||||
|
|
CH |
3 |
|||
|
|
|
|
CH |
||
|
|
3 |
|
|
||
|
|
|
|
|
3 |
2. Присоединение гидросульфита натрия. Из кетонов в эту реакцию вступают только метилкетоны:
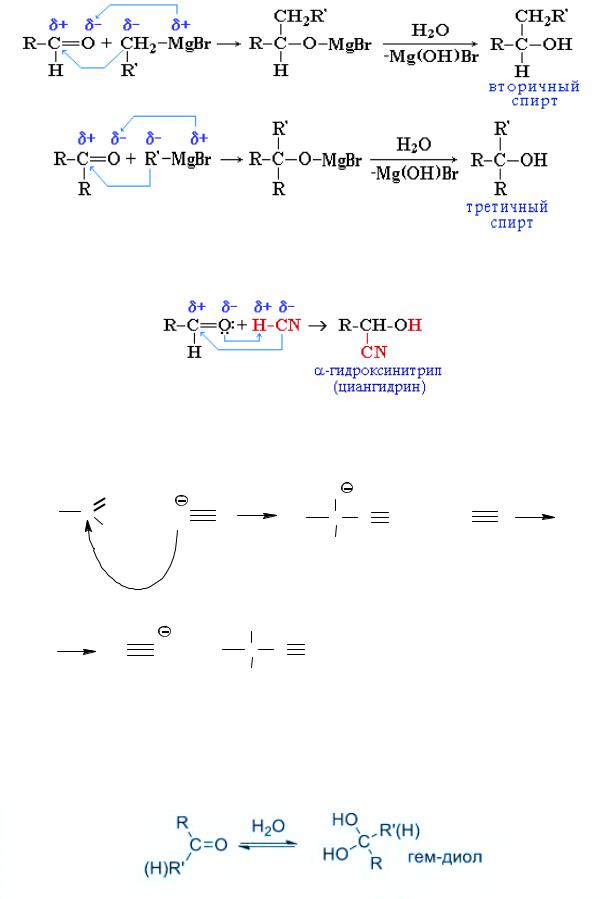
3. Реакции с С-нуклеофилами: реактивами Гриньяра, циановодородом.
А) Взаимодействие с реактивами Гриньяра – металлорганических соединений типа R-Mg-X , где Х= галоген:
506
Б) Присоединение синильной кислоты. Присоединение синильной кислоты ведет к образованию α-оксинитрилов:
Механизм реакции. Эта реакция начинается с атаки атома углерода карбонильной группы цианид-ионом, поскольку реакция ускоряется в присутствии едкого кали или цианистого калия:
|
|
O |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
Ñ |
|
+ |
Ñ |
N |
R |
Ñ |
C |
N |
+ HÑ |
N |
|
|
H |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
OH |
|
|
о ксин итрил |
|
|
+ |
|
|
|
|
|
N |
Ñ |
R |
Ñ |
C |
N |
èëè |
|
|
|
|
|
H |
|
|
циан гидрин |
|
|
|
|
|
|
|
4. Реакции с гетеронуклеофилами: присоединение воды и спиртов, образование ацеталей.
А) Присоединение воды. Некоторые альдегиды и кетоны практически полностью присоединяют воду, образуя гем-диолы:
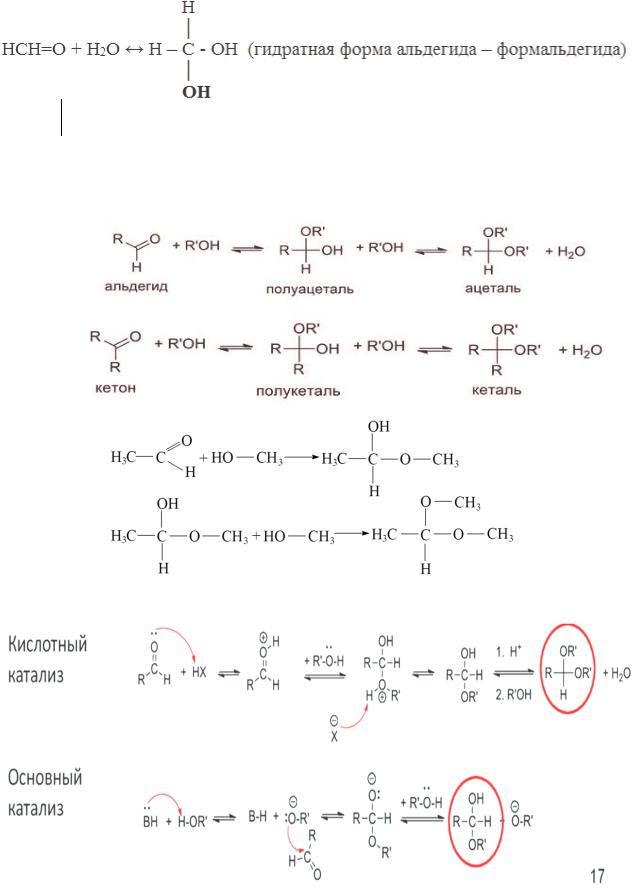
508
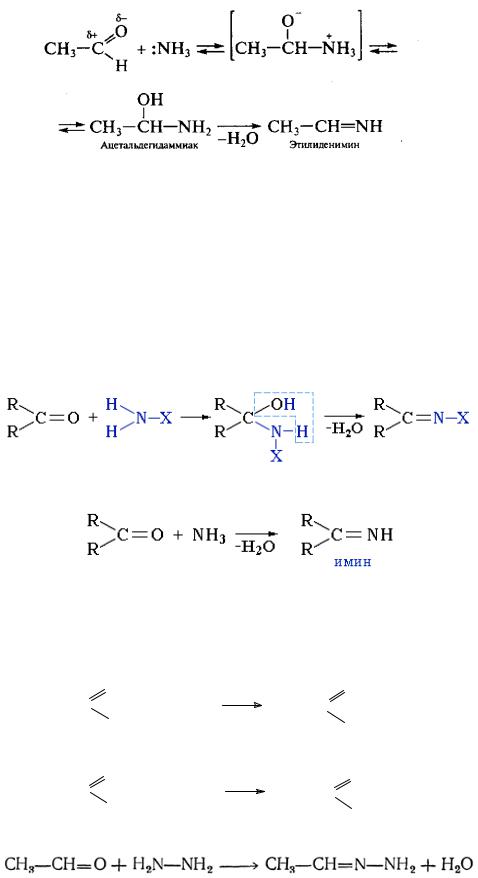
с аммиаком с образованием альдегидоаммиаков. Последние малоустойчивы и легко дегидратируются, превращаясь в альдимины:
Кетоны с аммиаком реагируют медленно и в достаточно жестких условиях, поэтому реакция с аммиаком позволяет различать альдегиды и кетоны.
Б) Присоединение производных аммиака. Соединения, содержащие аминогруппу, реагируют с альдегидами и кетонами с образованием разнообразных производных. В случае первичных аминов получают кетимины и альдимины, которые являются промежуточными продуктами в общем способе получения вторичных аминов из альдегидов и кетонов (восстановительное аминирование):
При взаимодействии с аммиаком образуются имины:
При взаимодействии альдегидов и кетонов с гидроксиламином (NH2OH) получают оксимы, а с гидразином (H2N—NH2) и его производными
– гидразоны.
H3C |
|
|
C |
O |
+ H N |
|
|
OH |
H C |
|
|
|
N |
|
|
OH |
||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
C |
|
+ H2O |
|||||||||||
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
||||||||||||||
|
|
|
|
H |
2 |
|
|
|
|
3 |
|
|
|
H |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
H3C |
|
|
C |
O |
+ H2N |
|
|
|
OH |
H3C |
|
|
N |
|
|
OH |
||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
C |
|
+ H2O |
||||||||
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
||||
509
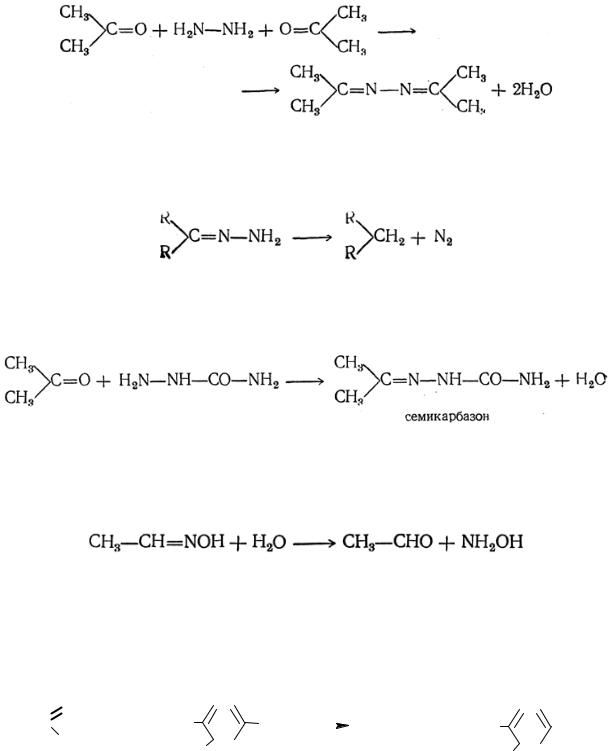
Гидразоны альдегидов и кетонов под влиянием твердых едких щелочей или алкоголятов подвергаются каталитическому разложению с выделением азота и замещением кислорода карбонильной группы исходного альдегида или кетона на водород:
С семикарбазидом (H2N—СО—NH—NH2) получаются соединения, называемые семикарбазонами, например:
При действии кислот на оксимы, гидразоны и семикарбазоны происходят реакции, обратные реакциям образования этих соединений, например:
Чаще для идентификации альдегидов и кетонов используют арилгидразины – это твердые вещества с характерными температурами плавления (например, 2,4-динитрофенилгидразон); используют для идентификации альдегидов и кетонов:
|
|
O |
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
Í |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
ÑÍ 3 |
|
Ñ |
H2N |
|
NH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
NO2 |
|
|
H C |
|
CH |
|
N |
|
NH |
|
|
|
|
|
NO |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
H |
|
|
O2N |
|
|
|
|
-Í 2O |
3 |
|
|
|
|
|
O2N |
|
|
|
|
|
|
2 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,4-дин итро ф ен илгидразо н
Имины – маслообразные или кристаллические вещества, обычно нерастворимые в воде и растворимые в органических растворителях. Несопряженные имины бесцветны, при сопряжении аминогруппы с ароматической системой или системой двойных связей – окрашены и используются в качестве красителей (азометиновые красители) и индикаторов, например, мурексид.
510
Перегруппировка Бекмана. Ароматические кетоны проявляют те же свойства, что и ароматические альдегиды. Особенностью являются оксимы несимметричных ароматических кетонов. В силу того, что в оксимах атомы углерода–азота соединены двойной связью, то возможна геометрическая изомерия. Причем в этом случае изомеры обозначаются «син» и «анти». Син-формой принято считать изомер, содержащий меньший радикал в цисположении по отношению к гидроксильной группе. По Е, Z-системе синформа будет рассматриваться как Е-изомер. Анти-формой принято считать изомер, содержащий меньший радикал в транс-положении по отношению к гидроксильной группе.
|
Ñ |
R |
Ñ |
|
R |
|
|
|
|
||
|
N |
|
N |
|
|
|
|
|
|
|
|
HO |
ñèí -êåòî êñèì , |
|
àí ò è-êåòî êñèì |
, |
OH |
|
|
||||
|
E-èçî ì åð |
|
Z-èçî ì åð |
|
|
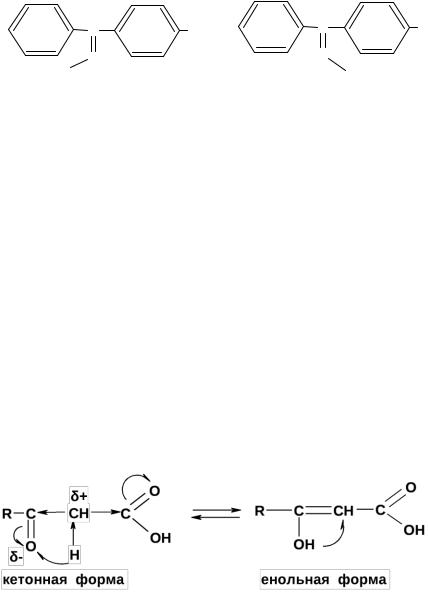
6. СН-кислотность карбонильных соединений и кето-енольная таутомерия.
Карбонильные соединения являются α-CH-кислотами вследствие резонансной стабилизации соответствующего аниона. Следствием CHкислотности является подвижность α-CH-протона – основная причина кетоенольной таутомерии, поскольку подвижный протон может обратимо мигрировать к неподеленной электронной паре кислорода.
Для альдегидов и кетонов, содержащих, по крайней мере, один α- водородный атом, возможна кето-енольная таутомерия.
Таутомерия – динамическая изомерия, при которой изомеры могут переходить друг в друга, находясь в растворе в состоянии подвижного равновесия. Таутомерия, связанная с переносом протона, называется прототропной. Кето-енольная таутомерия – частный случай прототропной таутомерии. Между кетонной и енольной формой осуществляется перенос протона. Атом водорода при α-атоме углерода обладает слабыми кислотными свойствами.
Таутомеры – структурные изомеры, которые находятся в быстром динамическим равновесии и не могут быть выделены в чистом виде.
511
При установившемся равновесии вещество содержит одновременно молекулы таутомеров в определенном соотношении. Процесс такого взаимопревращения таутомеров называется таутомеризацией.
Процесс перехода кето-формы в енольную называют енолизацией. Склонность к таутомеризации обусловлена тремя причинами:
1)электроноакцепторным характером карбонильной группы, способствующим увеличению кислотности соседних связей С-Н;
2)определенной основностью карбонильного кислорода, к которому перескакивает протон;
3)резонансной стабилизацией енола, в котором неподеленная электронная пара гидроксила сопряжена с двойной связью С=С.
Для кетонов кето-форма гораздо более стабильна, чем изомерная енольная форма. Иначе говоря, это означает, что енол является на несколько порядков более сильной О-Н кислотой, чем кетон С-H кислотой.
7. Реакции енольных форм: α-галогенирование, галоформное расщепление, изотопный обмен водорода.
А) Галогенирование карбонильных соединений. Альдегиды и кетоны, имеющие подвижный атом водорода при α-углеродном атоме, легко вступают в реакции галогенирования в присутствии как кислот, так и оснований:
Необходимо отметить, что скорость реакции галогенирования альдегидов и кетонов не зависит от вида галогена. Она зависит только от вида и количества катализатора, определяющего лимитирующую стадию процесса — образование енола или енолят-аниона, которые затем быстро атакуются реагентом.
Б) Галоформное расщепление, изотопный обмен водорода.
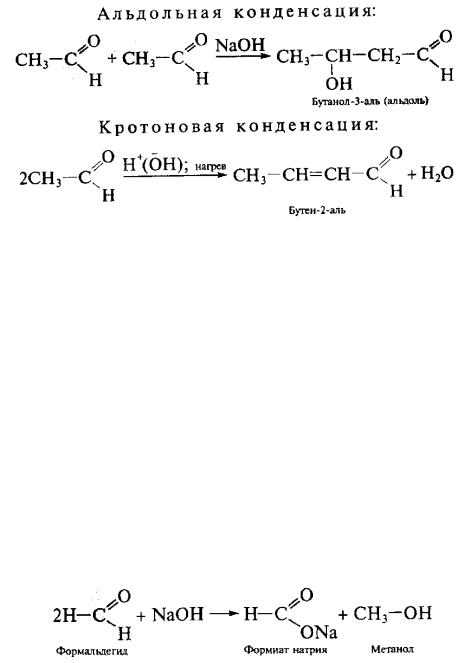
8. Альдольно-кротоновая конденсация, кислотный и основный катализ. Альдоли и α, β-непредельные карбонильные соединения.
А) Альдольная и кротоновая конденсации. Реакция конденсации приводит к удлинению углеродного скелета за счет возникновения новой углерод-углеродной связи и, сопровождается выделением воды или другого низкомолекулярного вещества.
512
В качестве примера приведем конденсацию уксусного альдегида под влиянием разбавленных щелочей (А.П. Бородин, 1863-1873; Ш.А. Вюрц), при которой в реакцию вступают две молекулы альдегида, одна реагирует своей карбонильной группой, а вторая – углеродным атомом в α-положении к карбонильной группе, содержащим подвижный атом водорода, по схеме:
Названия этих синтезов происходят от конечных продуктов реакций. В первом случае образуется альдегидоспирт (альдоль), во втором – непредельный кротоновый альдегид.
Аналогично в реакцию альдольной конденсации вступают и кетоны. Только в случае кетонов реакция протекает значительно труднее.
Б) Альдоли и α, β-непредельные карбонильные соединения.
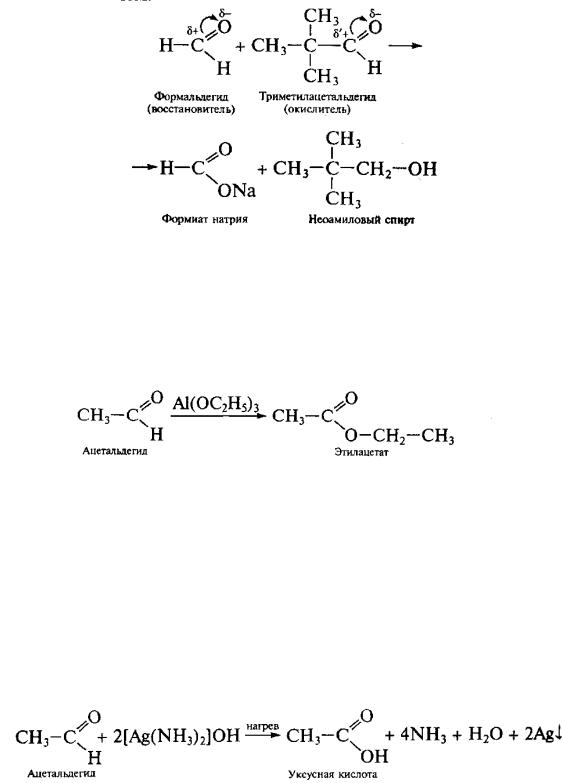
9. Взаимодействие неенолизирующихся альдегидов со щелочами (реакция Канниццаро). Бензоиновая конденсация.
А) Реакция Канниццаро. Реакция представляет собой окислительновосстановительное диспропорционирование альдегидов с действием концентрированного водного или водно-спиртового раствора щелочи при слабом нагревании в присутствии катализаторов (Со, Ni, Ag, Си или их оксидов), сопровождающееся образованием первичных спиртов и солей карбоновых кислот. В эту реакцию вступают альдегиды, в молекулах которых отсутствуют подвижные α-водородные атомы.
При наличии подвижных α-водородных атомов протекает, как правило, реакция альдольной конденсации.
Реакция Канниццаро дает хорошие результаты при взаимодействии двух альдегидов, отличающихся полярностью карбонильных групп. Тогда один из них является окислителем, а другой – восстановителем.
513
Б) Реакция Тищенко (сложноэфирная конденсация альдегидов).
Это реакция диспропорционирования альдегидов, содержащих α-водородные атомы, под действием алкоголят алюминия с образованием сложных эфиров.
Реакцию Тищенко проводят в безводной среде (иногда в инертном органическом растворителе) при комнатной температуре.
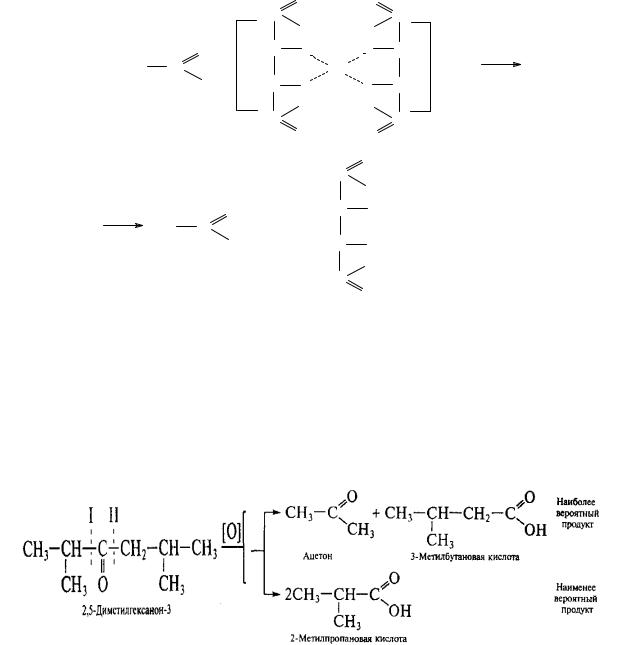
10. Реакции окисления карбонильных соединений. В отличие от кетонов альдегиды легко окисляются кислородом воздуха до карбоновых кислот, поэтому их хранят в плотно закупоренных сосудах. В качестве окислителей применяют аммиачный раствор гидроксида серебра, реактив Фелинга или перманганат калия. Эти реакции находят широкое применение в аналитической химии, так как позволяют различить альдегиды и кетоны.
Реакция «серебряного зеркала». При легком нагревании аммиачного раствора гидроксида серебра с альдегидом происходит его окисление до карбоновой кислоты с образованием свободного металлического серебра, которое ровным слоем ложится на предварительно обезжиренную внутреннюю поверхность пробирки:
Для альдегидов также характерно взаимодействие с реактивом Фелинга, представляющим собой водно-щелочной раствор комплексной соли, образующейся из гидроксида меди (II) и натрий-калиевой соли винной кислоты. При нагревании альдегидов с этим реактивом происходит его окисление до карбоновой кислоты, а гидроксид меди(II) восстанавливается до гидроксида меди (I):
514
R–CH=O + 2Cu(OH)2 → RCOOH + Cu2O + 2H2O
|
|
|
C |
O |
O |
C |
|
|
|
|
|
|
ONa |
KO |
|
2+ |
|
||
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
||
|
|
O |
HC |
OH |
HO HC |
2OH |
- |
||
H C |
C |
+ |
Cu |
|
|
||||
|
|
|
|
||||||
3 |
H |
|
|
|
|
|
|
||
|
|
HC |
OH |
HO |
HC |
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
C |
OK Na O |
C |
|
|
|
|
|
|
|
O |
O |
|
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
C |
O |
|
|
|
|
|
|
|
|
ONa |
|
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
O |
HC |
OH |
|
|
|
|
|
H C |
|
|
|
|
|
|
|
|
|
C |
+ Cu O + |
|
+ |
H O |
|
|||
|
3 |
|
OH |
2 |
|
|
2 |
|
|
|
|
|
HC |
OH |
|
|
|
||
|
|
|
|
C |
OK |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
Окисление кетонов идет с большим трудом и в жестких условиях (щелочной раствор перманганата калия, горячая концентрированная азотная кислота, хромовая смесь — дихромат калия в серной кислоте). Оно осуществляется с разрывом углеродной цепи по обе стороны от карбонильной группы, и приводит в общем случае к образованию четырех карбоновых кислот, однако окисление наиболее вероятно у наименее гидрогенизированного атома углерода (правило Попова):
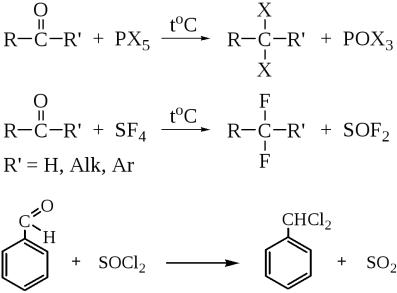
11. Реакции замещения кислорода карбонильной группы галогеном
Под действием различных галогенсодержащих реагентов атом кислорода в карбонильной группе может замещаться на галоген. Продуктами реакции являются гем-дигалогенпроизводные. Эта реакция осуществляется под действием галогенидов фосфора или серы при нагревании.
515
Такие дигалогенопроизводные, реагируя с водой в присутствии кислот, способны опять давать исходные альдегиды или кетоны.
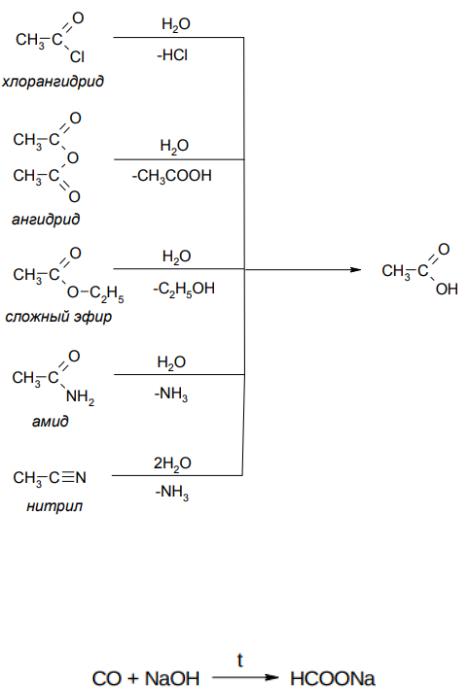
3.6.6 Карбоновые кислоты и их производные
Карбоновые кислоты — производные углеводородов, содержащие карбоксильную группу -СООН.
Карбоновые кислоты и их производные чрезвычайно важны в биологической химии, поскольку все важнейшие биохимические пути связаны с превращениями карбоновых кислот и их производных.
Многие карбоновые кислоты были выделены из жиров, поэтому карбоновые кислоты также называются жирными кислотами.
Номенклатура
Название «кислота» образовано от слова «кислый», а слово «карбоновая» восходит к латинскому слову carboneum — углерод.
По номенклатуре ИЮПАК названия кислот образуют, используя названия соответствующих углеводородов, прибавляя окончание «-овая» и слово «кислота». Например, муравьиная кислота НСООН содержит один атом углерода, следовательно, она является производной метана. Ее международное название — метановая кислота.
Уксусная кислота СН3СООН содержит два атома углерода — как и углеводород этан, следовательно, ее название — этановая кислота. Однако на практике чаще используются тривиальные названия (таблица 3.6.6.1).
516
Таблица 3.6.6.1 – Названия карбоновых кислот
Число |
атомов |
Название |
|
Название соли |
Тривиальное |
Тривиальное |
Этимология |
|
|
С |
|
кислоты |
по |
по ИЮПАК |
название |
название соли |
тривиального |
|
|
|
|
ИЮПАК |
|
|
кислоты |
|
названия |
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
метановая |
|
метаноат |
муравьиная |
формиат |
Лат. |
Formica- |
|
|
|
|
|
|
|
|
муравей |
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
этановая |
|
этаноат |
уксусная |
ацетат |
Лат. Acetum |
- |
|
|
|
|
|
|
|
|
уксус |
|
|
|
|
|
|
|
|
|
|
||
3 |
|
пропановая |
|
пропаноат |
Пропионовая |
пропионат |
гр. - первый, - |
||
|
|
|
|
|
|
|
жир |
|
|
|
|
|
|
|
|
|
|
||
4 |
|
бутановая |
|
бутаноат |
масляная |
бутират |
Лат. Butyrum- |
||
|
|
|
|
|
|
|
масло |
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
пентановая |
|
пентаноат |
валерьяновая |
валерат |
Лат. |
Valeriana |
|
|
|
|
|
|
|
|
- валериана, |
||
|
|
|
|
|
|
|
(valere - быть |
||
|
|
|
|
|
|
|
сильным) |
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
гексановая |
|
гексаноат |
капронат |
капронат |
Лат. |
Caper |
- |
|
|
|
|
|
|
|
коза |
|
|
|
|
|
|
|
|
|
|
|
|
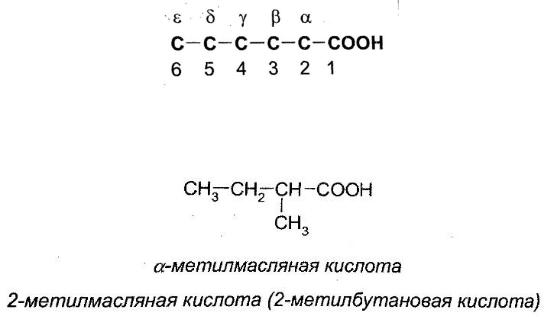
Для карбоновых кислот используются две системы нумерации — по рациональной номенклатуре (греческими буквами) и IUPAC (арабскими числами):
Обратите внимание, что в системе ИЮПАК первый номер получает атом углерода группы СООН, а в рациональной системе греческой буквой обозначается атом углерода, присоединенный к СООН.
Названия солям обычно дают, используя тривиальные названия: CH3COONa – ацетат натрия, (CH3CH2COO)2Ca – пропионат кальция.
В биологической химии названия соли используется для обозначения всей совокупности различных молекулярных и ионных форм кислоты.
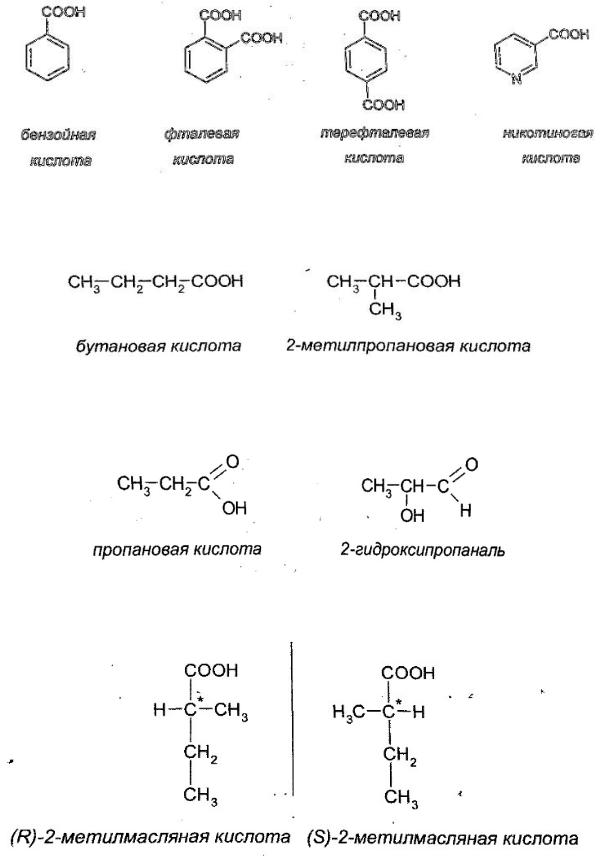
Для простейших ароматических кислот в основном используются тривиальные названия:


519
В основном увеличение температуры кипения происходит за счет образования очень прочных межмолекулярных водных связей между двумя молекулами. Карбоновые кислоты в твердом виде, в жидкой и даже в газовой фазе существуют в виде димеров:
Диамер — структура, образованная объединениями двух молекул.
В твердом виде практически все карбоновые кислоты существуют в виде димеров, например, пропионовая кислота:
Интересно, что муравьиная и уксусная кислота в твердом виде, как и спирты, образуют цепочки, а не димеры:
Первые члены гомологического ряда карбоновых кислот обладают резким запахом, средние — прогорклым, неприятным, например, масляная кислота пахнет потом, высшие карбоновые кислоты вследствие нелетучести лишены запаха.
Карбоновые кислоты, как правильно, не ядовиты, однако прием внутрь концентрированных растворов (например, уксусной эссенции) вызывает тяжелые ожоги. Нежелательно попадание этих растворов на кожу и тем более внутрь.
Строение карбоксильной группы и карбоксилат-иона
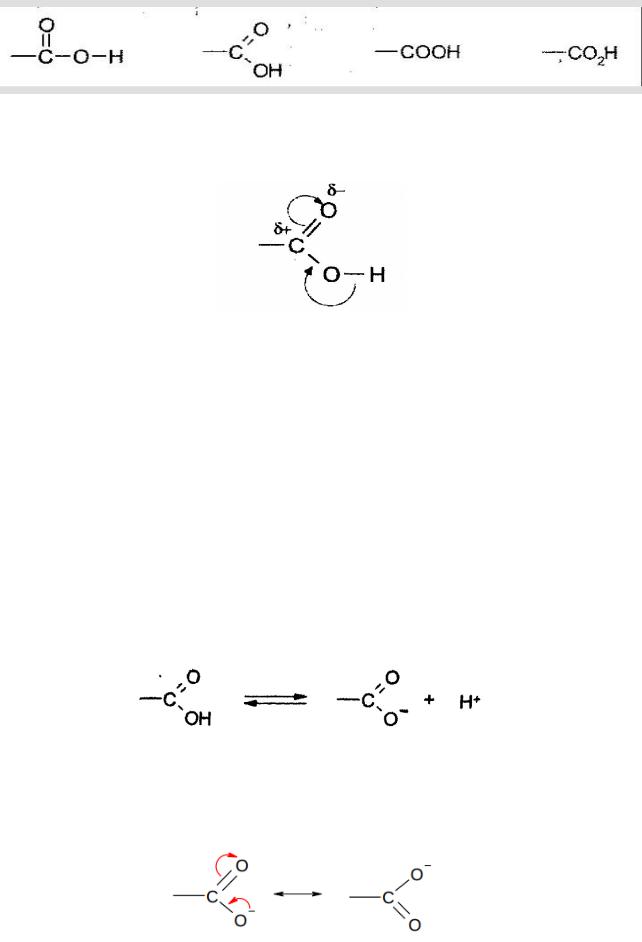
Карбоновые кислоты содержат карбоксильную группу, которая может изображаться по-разному:
521
которыми стоит обоюдоострая стрелка. Это означает, что в реальности карбоксилат-анион представляет собой гибрид этих двух структур: двойная связь С=О не является двойной и отрицательный заряд не сосредоточен только на одном атоме кислорода. То есть строение карбоксилат-аниона в реальности больше соответствует структуре, получающейся при наложении двух крайних резонансных формул:
Карбоновые кислоты являются слабыми кислотами. При концентрации 0,1 моль/л СН3СООН диссоциирована примерно в 100 раз хуже, чем HСl. Однако, карбоновые кислоты сильнее, чем угольная кислота, синильная, сероводородная. Количественной характеристикой силы кислоты является константа диссоциации:
Если кислота является более сильной, то это означает, что она лучше диссоциирует и тем больше К.
Наиболее сильной среди карбоновых кислот является муравьиная (метановая) кислота. При введении электродонорного углеводородного радикала кислотность уменьшается, поэтому уксусная кислота слабее, чем муравьиная.
Итак, карбоновые кислоты проявляют кислотные свойства, индикаторы меняют в них свою окраску, например, лакмус в растворе уксусной кислоты становится красным.
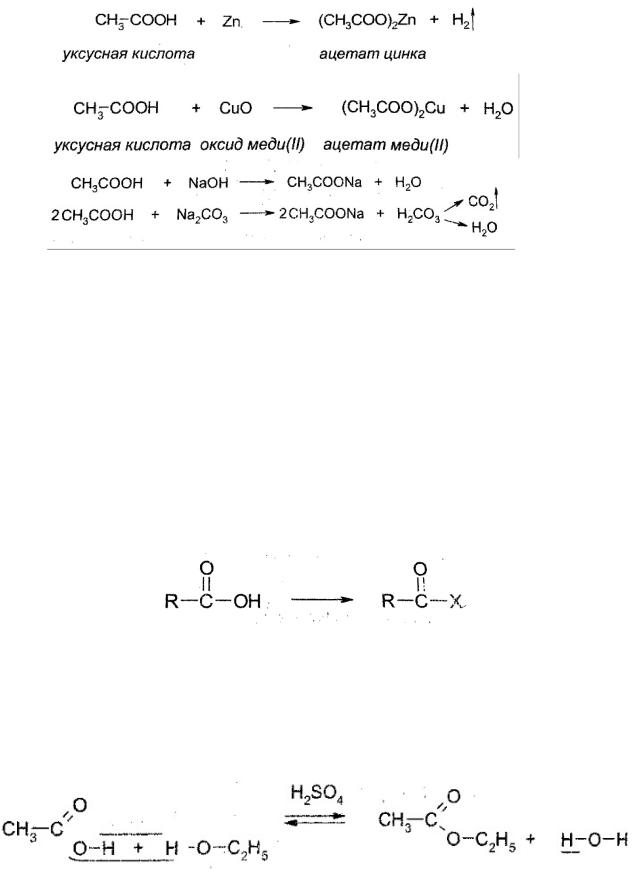
Карбоновые кислоты проявляют все общие свойства кислот, взаимодействуют с металлами, стоящими до водорода, реагируют с основными и амфотерными оксидами, амфотерными гидроксидами, щелочами, солями более слабых кислот:
522
Как известно, соли, образованные слабыми кислотами, подвергаются гидролизу. Это относится и к солям слабых карбоновых кислот. Водные растворы натриевх солей имеют щелочную среду:
СН3СООNa+H2O=CH3COOH+NaOH
По этой же причине мыла также дают щелочную среду:
C15H31COONa+H2O=C15H31COOH+NaOH
Мыла — натриевые или калиевые соли высших карбоновых кислот: в основном пальмитиновой C15H31COOH и стеариновой C17H35COOH.
2. Нуклеофильное ацильное замещение
Гидроксигруппа в карбоксильной группе может замещаться с образованием производных карбоновых кислот:
2.1. Образование сложных эфиров — этерификация по Фишеру
Термин «этерификация» (нем. Etherifikation) происходит от слова — эфир. Этерификация — это образование эфиров. Как правило, этот термин относится только к образованию сложных эфиров.
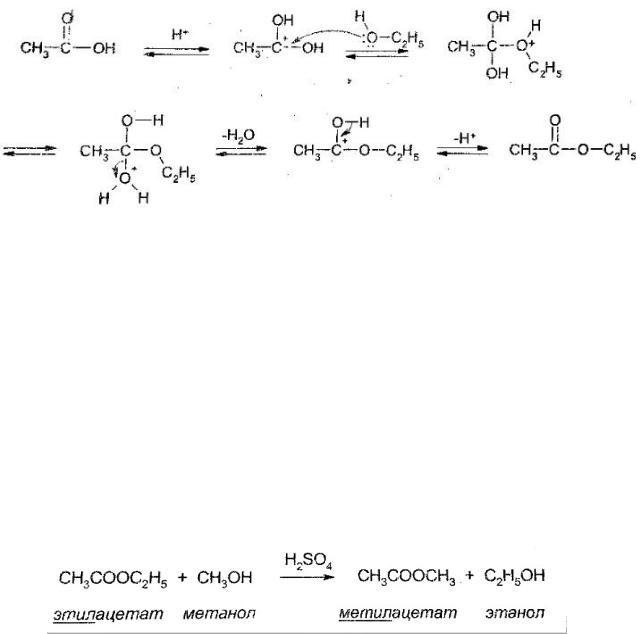
Итак, карбоновые кислоты реагируют со спиртами в присутствии катализатора — серной кислоты с образованием сложных эфиров:
Роль серной кислоты состоит в протонировании карбоновой кислоты, в увеличении положительного заряда на карбонильном атоме углерода, а также в связывании образующейся воды.
523
Механизм реакции этерификации:
На первом этапе серная кислота протонирует молекулу карбоновой кислоты, на атоме углерода которой возникает значительный положительный заряд.
На втором этапе молекула спирта (нуклеофил) атакует положительно заряженный атом углерода, образуя продукт присоединения, который отщепляет воду, образуя протонированный сложный эфир,
На третьем этапе от протонированного сложного эфира отщепляется протон (регенерация катализатора) с образованием сложного эфира.
Механизм гидролиза сложного эфира обратен. То есть если переписать механизм образования сложного эфира в обратном направлении, то получится механизм гидролиза сложного эфира. Аналогичный механизм наблюдается и в случае реакции переэтерификации: при нагревании сложного эфира с избытком другого спирта в присутствии серной кислоты идет замещение остатка спирта на остаток другого спирта:
Образование и гидролиз сложных эфиров является реакцией нуклеофильного замещения и описывается как механизм ААС2
(бимолекулярная реакция катализируемая кислотами (лат. Acidum - кислота), с расщеплением связи С-О, где атом углерода принадлежит ацильному остатку).
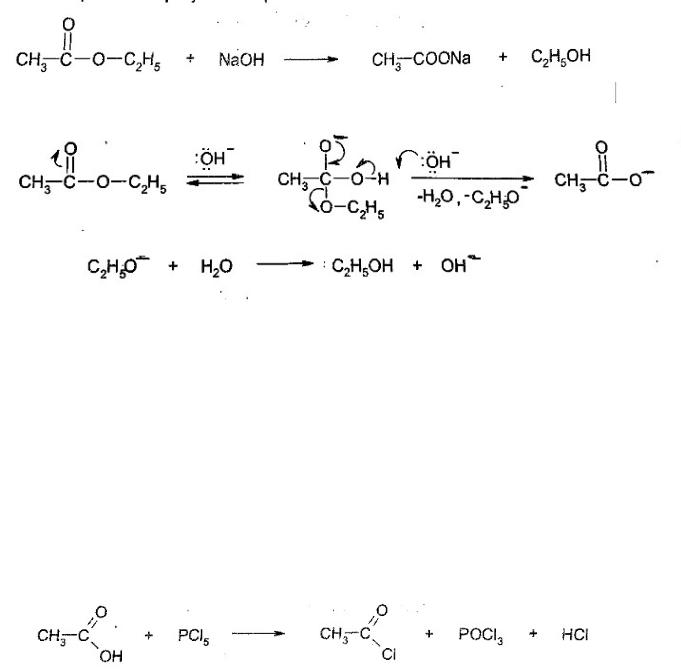
Сложные эфиры отлично гидролизуются под действием оснований. Щелочной гидролиз часто называется омылением. Например, при нагревании сложного эфира с щелочью образуется спирт и соль:
524
Механизм гидролиза, катализируемого основаниями:
Гидроксид-ион присоединяется к молекуле сложного эфира, образуя тетраэдрический интермедиат. Далее идет внутримолекулярное нуклеофильное замещение, в ходе которого высвобождается этоксид-ион. Он отщепляет протон от молекулы воды, превращаясь в спирт и регенерируя катализатор — гидроксид-ион.
В данном случае механизм этого нуклеофильного ацильного замещения описывается как ВАС2 (бимолекулярная реакция, катализируемая основаниями (лат. Basis - основания), с расщеплением С-О, где атом углерода принадлежит ацильному остатку).
2.2. Образование галогенангидридов
При взаимодействии карбоновой кислоты с галогенидами фосфора, образуются очень важные производные — галогенангидриды.
Названия галогенангидридов обычно строятся таким образом: ацил+галогенид. Поэтому, образовавшееся в ходе приведенной реакции вещество имеет следующее название — ацетилхлорид (или этаноилхлорид).
Атомы галогена в галогенангидридах могут легко замещаться при действии различных нуклеофилов, поэтому галогенангидриды являются основой для синтеза разнообразных производных карбоновых кислот.
3. Галогенирование карбоновых кислот
При действии хлора или брома на карбоновую кислоту в присутствии фосфора при нагревании (100°С), происходит замещение атома водорода на галоген в α-положении:
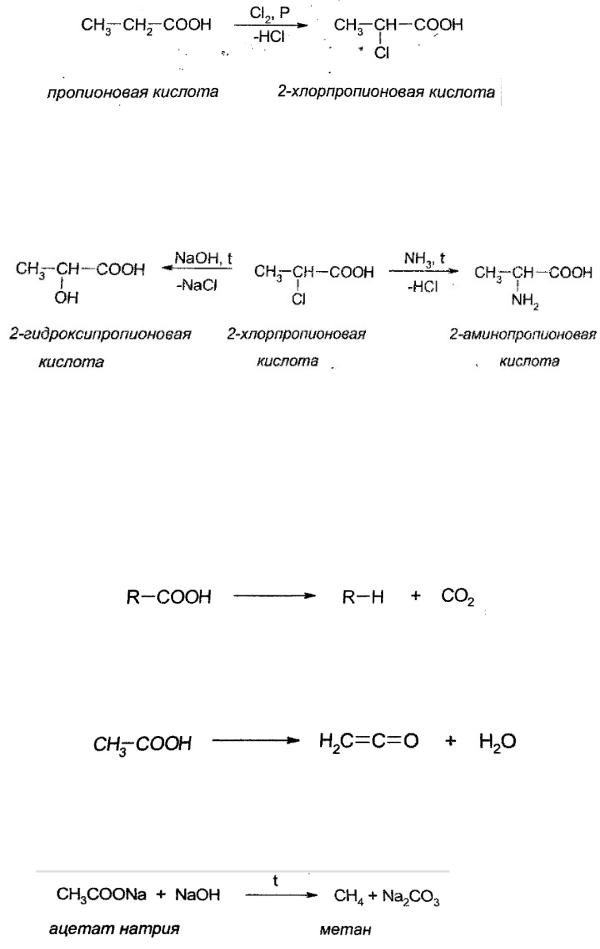
525
Данная реакция идет по механизму электрофильного замещения (SE)
с промежуточным образованием галогеноангидрида. Реакция имеет очень большое синтетическое значение: используя эту реакцию, получают галогенопроизводные карбоновых кислот, из которых можно получить аминокислоты, гидроксикислоты и т.д.
Реакция замещения галогена в α-галогенкарбоновых кислотах на гидроксигруппу и аминогруппу являются реакциями нуклеофильного
замещения (SN).
4. Декарбоксилирование Декарбоксилирование – это процесс потери группы СООН,
превращаясь в другое соединение.
В самом простом случае кислота теряет молекулу СО2, превращаясь в углеводород:
Однако такой процесс идет только с кислотами с электроноакцепторными заместителями. При нагревании (600-700 ᴏС) уксусной кислоты, она не декарбоксилируется, а, отщепляя воду, превращается в кетан:
Однако, при нагревании солей с щелочами, декарбоксилирование возможно; при этом образуется не СО2, а карбонат — соль угольной кислоты. Итак, из солей карбоновых кислот сплавлением со щелочью получают углеводороды (реакция Дюма):
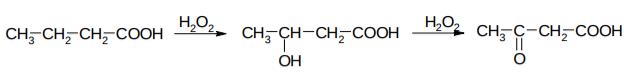
526
Если при декарбоксилировании происходит еще и окисление кислоты, то такое декарбоксилирование называется окислительным.
5. Окисление и восстановление карбоновых кислот
Все карбоновые кислоты горят с образованием углекислого газа и воды (например, горение стеариновой и пальмитиновой кислот наблюдается при горении стеариновой свечи).
В организме карбоновые кислоты окисляются в основном за счет т.н. β- окисления. Кроме того, in vivo встречается также α- и ω-окисление. Подробно β-окисление будет изучаться в курсе биохимии. In vitro некоторые аналогичные реакции β-окисления можно осуществить с помощью 3% перекиси водорода. Например, масляная кислота превращается в β- гидроксимасляную, а последняя окисляется до β-оксомасляной (ацетоуксусной) кислоты:
Карбоксильная группа восстанавливается с большим трудом. Для того, чтобы восстановить карбоновую кислоту до углеводорода, требуется длительное кипячение с HI в присутствии фосфора.
Прямое восстановление карбоновых кислот до спиртов водородом достигается при использовании высоких давлений и катализатора (Cu, Ni, Co, Zn-Cr-Cu-Cd).
Получение карбоновых кислот 1. Из природных источников
Исторически первым способом получения карбоновых кислот было их получение из природных источников. Например, муравьиную кислоту можно выделить из муравьёв, которые используют 70% муравьиную кислоту в качестве оружия. Также её можно обнаружить в хвое, крапиве, гусеницах, пчёлах.
При скисании вина образовьвался уксус, при перегонке которого можно получить уксусную кислоту. При замораживании уксусной кислоты, в первую очередь кристаллизуется твёрдая уксусная кислота (т. пл. 16,8 °С), которую отделяют от незамерзшей разбавленной уксусной кислоты. В результате можно легко получить 100% уксусную кислоту, которая называется «ледяной», несмотря на то, что она может быть и комнатной температуры. Название «ледяная уксусная кислота» отражает способ получения 100% уксусной кислоты, а не её температуру (отсюда возможна такая фраза «кипящая ледяная уксусная кислота"). В настоящее время уксусную кислоту для пищевых целей получают главным образом скисанием перебродивших соков (виноградного или яблочного). В промышленных масштабах используется неполное окисление бутана кислородом воздуха.
2. Окисление углеводородов
Низшие алканы, содержащиеся в попутных нефтяных газах, могут быть каталитически окислены до уксусной кислоты, причем в качестве примеси
527
получается и муравьиная кислота.
Высшие алканы под действием кислорода воздуха в жидкой фазе при нагревании превращаются в смесь карбоновых кислот со средней длиной цепи С12-С18, которые используются для получения моющих средств и ПАВ. Обыкновенный продажный парафин можно окислить до карбоновых кислот, пропуская через него воздух при 100-160 .
3. Гидролиз производных карбоновых кислот
Все производные карбоновых кислот гидролизуются до карбоновых кислот в условиях кислотного или основного катализа:
Промышленное значение имеет гидролиз жиров – сложных эфиров высших жирных кислот и трѐхатомного спирта глицерина (см. далее).
4. Вытеснение из солей сильными кислотами
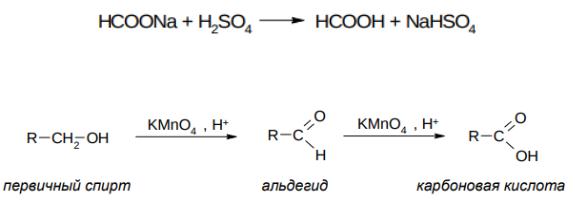
В промышленности муравьиную кислоту получают, пропуская угарный газ через расплавленный NaOH. При этом образуется формиат натрия – муравьинокислый натрий:
Из образующегося формиата натрия муравьиную кислоту выделяют действием сильных кислот:
528
5. Окисление альдегидов и спиртов
Первичные спирты легко окисляются в альдегиды, а альдегиды в карбоновые кислоты.
Аналогичным образом ферментативно метаболизируется этиловый спирт в организме в печени: вначале этанол окисляется до ацетальдегида и далее до уксусной кислоты, которая «сгорает» в цикле Кребса.
Здесь мы привели лишь несколько способов получения карбоновых кислот. Мы не упомянули такие методы, как синтез с использованием реактивов Гриньяра, метод Реппе, гидролиз 1,1,1-тригалогенпроизводных, присоединение угарного газа к спиртам, синтез Арндта-Эйстерта и др.
ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ ПРЕДЕЛЬНЫХ ОДНООСНЫХ КИСЛОТ
Муравьиная кислота Н-СООН. Безводная муравьиная кислота – бесцветная жидкость с резким запахом. Технический продукт представляет собой нераздельно кипящую смесь с водой (Ткип. 107,3 оС), содержащую 77,5 % кислоты.
Как уже указано, муравьиная кислота в отличии от других кислот содержит в соединении с карбоксилом не углеводородный радикал, а водород, и из ее формулы видно, что в ней имеется как бы альдегидная группа >С=О, соединенная с гидроксилом. Поэтому, подобно альдегидам муравьиная кислота является сильным восстановителем и окисляется до угольной кислоты, разлагающейся с образованием СО2 и Н2О. В частности, она восстанавливает окись серебра (реакция серебряного зеркала):
Н-СО-ОН + Ag2O (NH4OH) → 2Ag + CO2 + H2O
Под действием серной кислоты при нагревании муравьиная к-та разлагается, образуя окись углерода и воды:
Н-СООН → СО + Н2О
Вприроде свободная муравьиная кислота встречается в выделениях муравьев, в соке крапивы, в поте животных.
Впромышленности муравьиную кислоту получают, пропуская окись углерода через нагретую щелочь:
529
NaOH + CO → HCOONa
Из образовавшейся соли муравьиную кислоту выделяют действием разбавленной серной кислоты:
H—COONa + H2SO4 → H—OOH + NaHSO4
Применяют муравьиную кислоту при крашении тканей, в качестве восстановителя, в различных органических синтезах.
Уксусная кислота СН3-СООН. Безводная уксусная кислота – бесцветная жидкость с характерным острым запахом, ее иначе называют ледяной уксусная кислотой, т.к. она замерзает уже около +16оС, образуя кристаллическую массу, напоминающую лед. Обычная крепкая уксусная кислота (уксусная эссенция) содержит 70–80 % кислоты.
Уксусная кислота – одно из наиболее давно известных органических веществ, в древности ее получали в виде уксуса при скисании вина. Она широко распространена в природе, содержится в выделениях животных, в растительных организмах, образуется в результате процессов брожения и гниения в кислом молоке, в сыре, при прогаркании масла и т.п. В промышленности уксусную кислоту получают следующими способами:
1. Из ацетилена (синтетическая уксусная кислота). Путем гидратации ацетилена по реакции Кучерова получают уксусный альдегид, последний кaталитически окисляют кислородом воздуха в уксусную кислоту. Схема процесса:
НОН, Hg |
O2, кат. |
СН≡СН → СН3—СН=О → СН3—СООН
Этим способом в настоящее время получают основное количество уксусной кислоты.
2. Из этилена (синтетическая уксусная кислота).
PdCl2, CuCl2 |
О2, кат. |
СН2=СН2 +О2 + Н2О → СН3—СОН |
→ СН3—СООН |
3. Уксуснокислым брожением жидкостей, содержащих этиловый спирт
(биохимическая уксусная кислота):
О2
СН3—СН2—ОН → СН3—СООН + Н2О
Этот процесс называется уксуснокислым брожением, он происходит при участии вырабатываемого «уксусным грибком» энзима и протекает сложным путем, через ряд промежуточных стадий. Этим способом в настоящее время получают сравнительно небольшое количество уксусной кислоты.
530
4.Сухой перегонкой дерева (лесохимическая уксусная кислота). При сухой перегонке дерева одним из продуктов является водный слой, содержащий до 10% уксусной кислоты. Его нейтрализуют известью, при этом образуется кальциевая соль уксусной кислоты (СН3СОО)2Са (древесный порошок), которую обрабатывают рассчитанным количеством серной кислоты, таким образом, выделяют концентрированную уксусную кислоту.
5.Окисление углеводородов нефти. Этот способ весьма перспективен и
впоследние годы приобретает все большее значение. Н.М. Эммануэлем предложен и разработан процесс прямого окисления бутана кислородом воздуха при 145 оС и 50 атм. по схеме:
Р, t, О2
2СН3-СН2-СН2-СН3 → 2СН3СООН + СН3-СО-СН2-СН3 + 2Н2О
Выход уксусной кислоты достигает 80 %, побочный продукт – метилэтилкетон. В качестве исходного можно использовать бутан из попутного нефтяного газа.
ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ I. Галогеноангидриды
Галогеноангидриды – это соединения, образующиеся при замещении группы OH в карбоновой кислоте на атом галогена (F, Cl, Br, I).
Химические свойства галогеноангидридов
Галогенид-ион является хорошей уходящей группой в реакциях нуклеофильного замещения, поэтому галогеноагнидриды очень активны в
реакциях нуклеофильного ацильного замещения (SNAcyl). Так, например,
галогеноангидриды легко взаимодействуют с различными нуклеофилами:
531
Вэтих реакциях ацильная группа формально замещает какой-либо атом
–атомы водорода в воде, спирте и аммиаке. Такое введение ацильной группы (RCO) называется ацилированием. В данном случае – когда вводится ацетильная группа (CH3CO) – такие реакции называются ацетилированием.
II. Ангидриды
Ангидриды – это соединения, образующиеся при отщеплении воды от кислот. В органической химии ангидридами называют производные карбоновых кислот, образующиеся при их межмолекулярной дегидратации (отщеплении воды). Молекула воды отщепляется от двух молекул карбоновой кислоты под действием сильных водоотнимающих веществ, например P2O5.
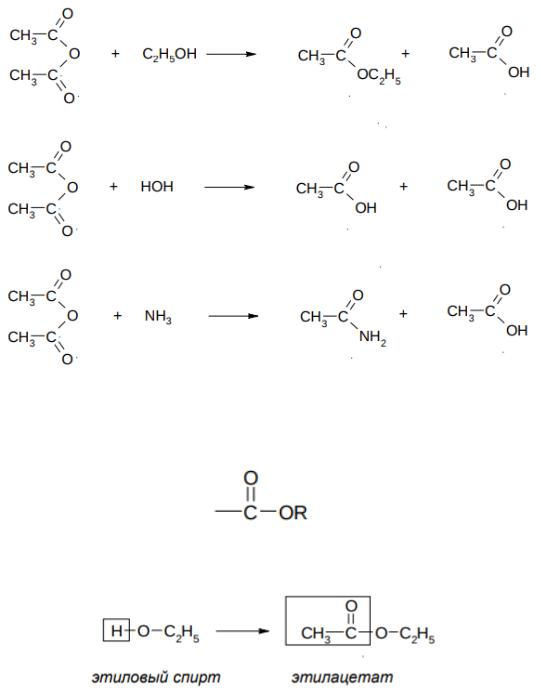

533
Для примера возьмем эфир, образованный уксусной кислотой и этиловым спиртом, который обычно называют этилацетатом:
Кроме того, этилацетат можно также назвать этиловым эфиром уксусной кислоты или уксусноэтиловым эфиром.
Эфиры карбоновых кислот часто встречаются в природе и природные эфиры можно подразделить на три группы:
1. Фруктовые эфиры – эфиры низших и средних карбоновых кислот с низшими и средними спиртами. Название этих эфиров обусловлено их приятным запахом. Некоторые из них являются составными частями эфирных масел, многие получаются синтетически и используются для придания запаха фруктовым сокам, лимонадам и т.д. Например, этилформиат (ромовая эссенция) используется для приготовления ромовой отдушки, изоамилацетат (грушевая эссенция) – для приготовления грушевого, ананасного и малинового масел. Изоамилизовалерат придает запах бананам и используется для приготовления яблочного, ананасного и персикового масел. Несмотря на то, что эфиры относительно безвредны и добавляются в небольших количествах, автор все-же считает, что все-таки лучше потреблять в пищу продукты без отдушек, и прочих добавок – это и вкуснее и полезнее.
2. Жиры – эфиры глицерина с высшими и средними жирными кислотами. Очень важные производные с точки зрения биологической химии. Кроме жиров существуют и другие боле сложные липиды (см. в теме Липиды).
3. Воска – эфиры высших одноатомных спиртов с высшими карбоновыми кислотами (см. в теме Липиды).
Химические свойства сложных эфиров 1. Ацильное нуклеофильное замещение
Сложные эфиры вступают в реакции нуклеофильного ацильного замещения: гидролиз, переэтерификация, аммонолиз и другие.
Гидролиз сложных эфиров ускоряется в присутствии каталитических количеств кислот:
Щелочной гидролиз сложных эфиров называется «омылением» и приводит к спирту и соли карбоновой кислоты.
Если нагревать сложный эфир этилацетат в метиловом спирте (или каком-то другом спирте) в присутствии каталитических количеств кислоты,
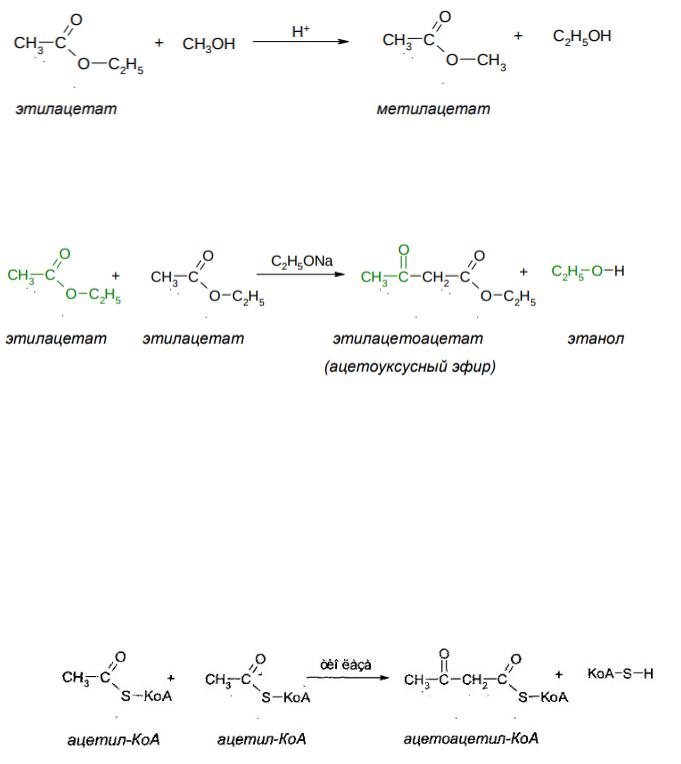
534
будет идти реакция переэтерификации: этилацетат превратится в метилацетат:
2. Сложноэфирная конденсация Кляйзена
Очень важной реакцией сложных эфиров является сложноэфирная конденсация Кляйзена: в щелочной среде сложные эфиры, содержащие атом водорода в α- положении, конденсируются с выделением спирта:
Таким образом получают ацетоуксусный эфир, являющийся сырьѐм для получения многих лекарственных веществ (напр., амидопирин, антипирин, акрихин, витамин В1)
Реакции, протекающие как сложноэфирная конденсация, хорошо известны в биологической химии. Однако, в организме задействованы сернистые аналоги сложных эфиров – эфиры кислот и коэнзима A, который кратко обозначается KoASH.
Первой стадией синтеза стероидов (в частности холестерина) и процесса кетогенеза, идущего особенно интенсивно при сахарном диабете, является конденсация двух молекул ацетил-КоА, представляющая собой сложноэфирную конденсацию:
Также аналогичные реакции (и обратные им) протекают при кетогенезе, синтезе терпенов и стероидов, β-окислении жирных кислот, и при метаболизме аминокислоты изолейцина.
Синтез сложных эфиров
Синтез сложных эфиров был рассмотрен в темах «Химические свойства карбоновых кислот», «Галогеноангидриды», «Ангидриды».
Полимерные сложные эфиры
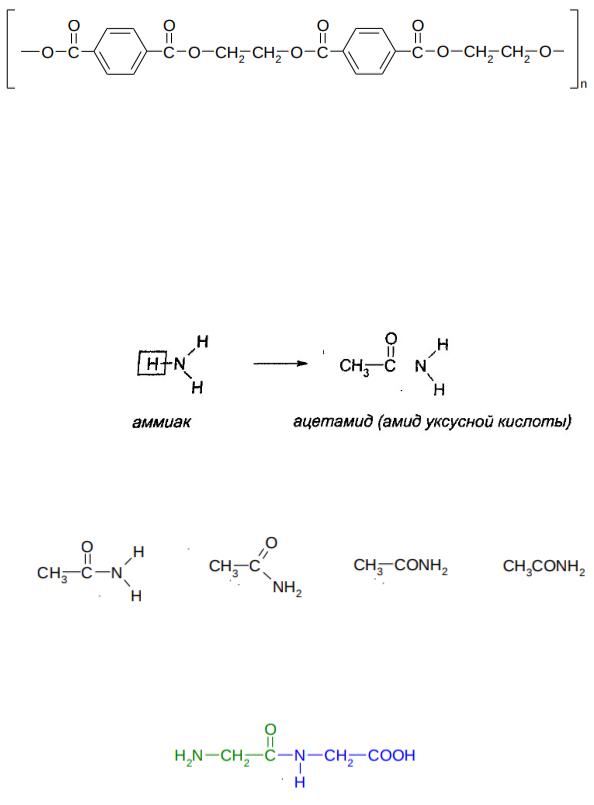
535
Примером синтетического полимерного сложного эфира является полиэтилентерефталат:
Полиэтилентерефталат, синтетический полимер, продукт поликонденсации этиленгликоля с терефталевой кислотой (или ее диметиловым эфиром); твердое бесцветное вещество. Полиэтилентерефталат перерабатывают главным образом в полиэфирные волокна – лавсан (дакрон, терилен и др. торговые названия), идущие на производство тканей. Сейчас полиэфирные волокна обычно называют полиэстером.
IV. Амиды
Амиды можно рассматривать как продукты замещения атома водорода в аммиаке (или аминах) на кислотный остаток.
Амиды можно рассматривать и как продукты замещения группы OH в кислотах на группу NH2.
Как и в случае многих других органических соединений, формула ацетамида может писаться различными способами:
Такие важные с биологической точки зрения вещества как белки и пептиды также являются амидами, в которых амидная связь (которая в биохимии называется пептидной связью) образуется между остатками двух аминокислот. Например, дипептид, образованный двумя остатками аминоуксусной кислоты:
Химические свойства амидов 1. Гидролиз
Гидролиз в данном случае представляет собой реакцию
нуклеофильного ацильного замещения (SNAcyl). Амиды легко
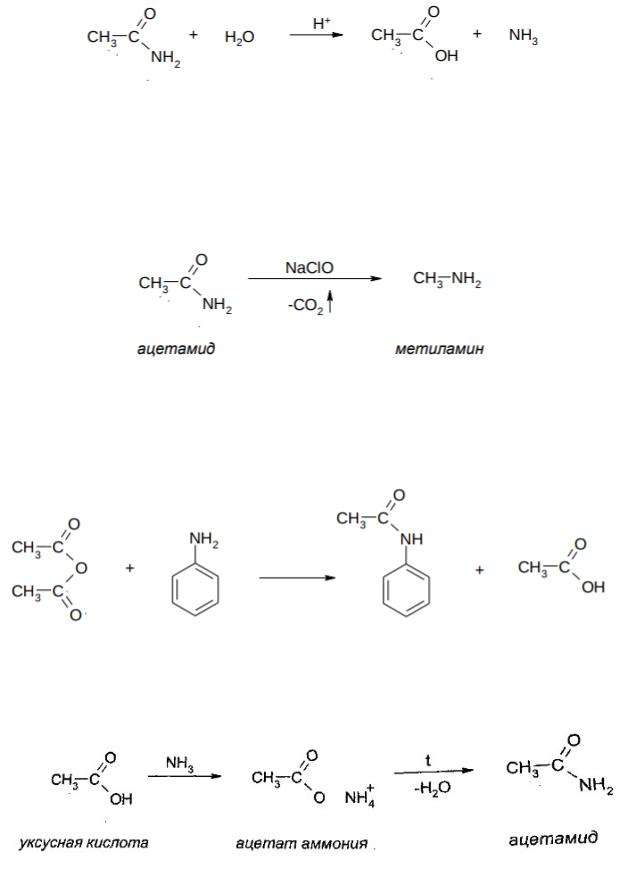
536
гидролизуются с образованием кислоты и аммиака при нагревании:
In vivo пептидные связи очень легко гидролизуются под действием ферментов, например, в желудке белки гидролизуются под действием фермента пепсина, в двенадцатиперстной кишке – под действием трипсина, химотрипсина и ряда других ферментов.
2. Реакция Гофмана
При действии на амид солей хлорноватистой кислоты при 50-80oC, идѐт образование аминов:
Получение амидов
Амиды обычно получают ацилированием аммиака или аминов, используя в качестве ацилирующих агентов галогеноангидриды или ангидриды кислот (см. темы «Галогеноангидриды», «Ангидриды»), например:
Непосредственно из аммиака (или аминов) и карбоновой кислоты амиды не получить по вполне понятной причине – кислота с аммиаком (или амидами) образует не амиды, а аммонийные соли. Однако, при нагревании аммонийных солей карбоновых кислот, образуются амиды:
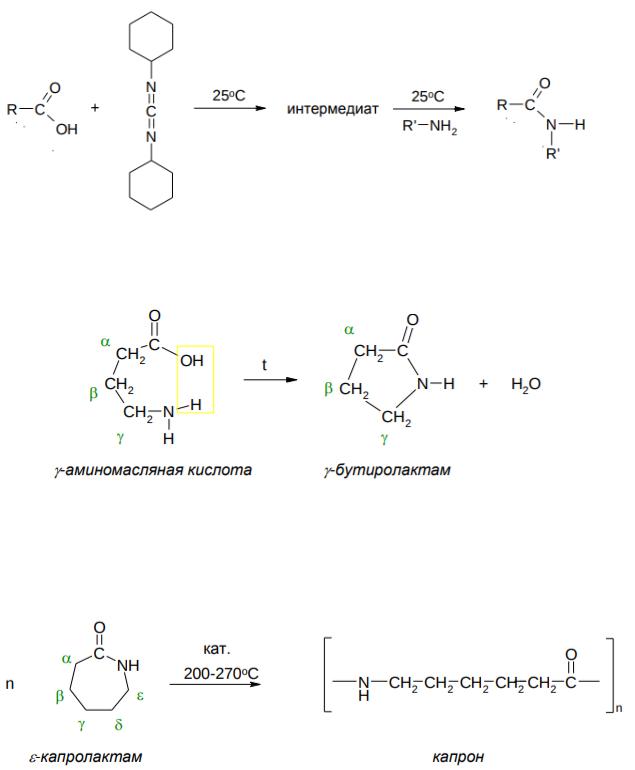
537
Однако, используя дициклогексилкарбодиимид (ДЦГК), можно получить амид непосредственно из карбоновой кислоты и амина, что используется в синтезе пептидов:
Лактамы и полимерные амиды – полиамиды
Циклические амиды называются лактамами. Обычно они легко образуются при нагревании γ- и σ-аминокислот (см. Аминокислоты):
Промышленно важный ε-капролактам – лактам ε-аминокапроновой кислоты – получают из фенола через ряд последовательных стадий. ε - капролактам – используется для получения синтетического полиамида –
капрона.
Синтетические полиамиды отличаются высокой механической прочностью, износостойкостью, химической устойчивостью.
Другой важный полиамид – найлон – получают сплавлением (180– 300oC) адипиновой кислоты и гексаметилендиамина:
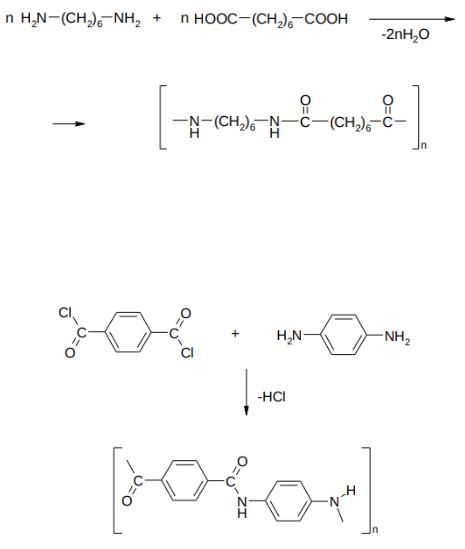
538
Из найлона и капрона получают полиамидное волокно, которое применяется в производстве тканей, трикотажа и т.д. Из нейлона делают струны для классической гитары и арфы.
Конденсация хлорангидрида терефталевой кислоты с п- фенилендиамином приводит к кевлару:
Прочность кевлара в пять раз выше, чем у стали и в 10 раз выше, чем у алюминия. Из кевлара изготавливают пуленепробиваемые жилеты, паруса для гоночных яхт, арматуру для пневматических шин и каски для гонщиков.
V. S-эфиры карбоновых кислот (сложные тиоэфиры)
Сложные тиоэфиры — органические соединения, содержащие функциональную группу R-S-CO-R1 и являющиеся сложными эфирами тиолов и карбоновых кислот.
Сложные тиоэфиры играют важную роль в биохимических процессах, наиболее известный представитель этого класса — ацетил-CoA. Другие примеры сложных тиоэфиров — малонил-CoA, ацетоацетил-CoA, пропионил-CoA и циннамоил-CoA. Сложные тиоэфиры — обычные интермедиаты во многих биохимических превращения, включая образование и распад жирных кислот и мевалоната — синтетического предшественника стероидов. Биосинтез лигнинов, составляющих значительную часть биомассы, протекает через образование сложного тиоэфира кофейной кислоты. Ацетогенез протекает через образование ацетил-CoA. Сложные тиоэфиры образуются в живых организмах в результате реакций эстерификации, причем АТФ играет роль дегидратирующего агента.
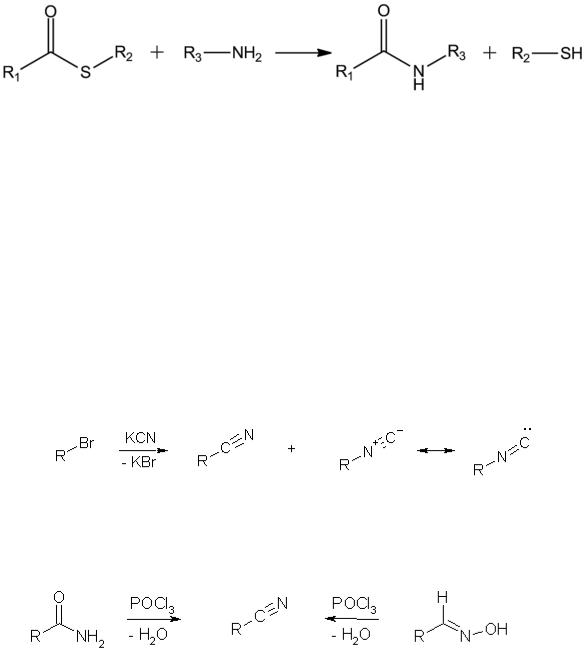
539
Способы получения
1. Важнейшим способом получения S-эфиры карбоновых кислот является конденсация тиолов и карбоновых кислот в присутствии водоотнимающих реагентов (например, N,N'-дициклогексилкарбодиимида
(DCC, ДЦК):
RSH + R1COОH → RSC(O)R1 + H2O
Химические свойства
1.Карбонильная группа сложных тиоэфиров реакционноспособна по отношению к атаке нуклеофилами (в меньшей степени по сравнению с хлорангидридами кислот). Так, сложные тиоэфиры реагирут с аминами с образованием амидов:
2.Ацилирование аминов сложными тиоэфирами:
CH3COSCH3 + CH3NH2 → CH3CONHCH3 + CH3SH.
VI. Нитрилы
Нитрилы — органические соединения общей формулы R—C≡N.
Группа —C≡N называется нитрильной группой.
Нитрилы также часто рассматривают как производные карбоновых кислот (продукты дегидратации амидов) и именуют как производные соответствующих карбоновых кислот, например, CH3C≡N — ацетонитрил (нитрил уксусной кислоты), C6H5CN — бензонитрил (нитрил бензойной кислоты).
Получение
1.При взаимодействии алкилгалогенидов с цианид-ионом:
2.Из других производных карбоновых кислот получение нитрилов
сводится к реакции дегидратации первичных амидов под действием P2O5, POCl3, уксусного ангидрида и других водоотнимающих средств:
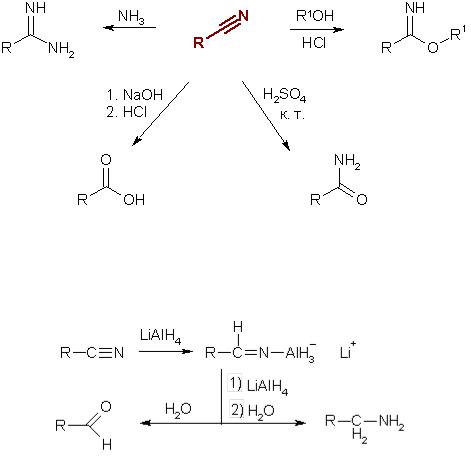
540
Химические свойства
Полярная тройная связь нитрилов способна присоединять нуклеофильные реагенты. Реагент присоединяется к атому углерода, однако электрофильность этого центра невысока, поэтому в большинстве случаев для успешного осуществления реакции необходимо активировать C≡N- группу протоном или кислотой Льюиса. В результате этих реакций образуются другие представители производных карбоновых кислот либо сами кислоты. Превращение, по сути, представляет собой реакцию ацилирования.
Нитрилы восстанавливаются до алкиламинов при действии алюмогидрида лития. Реакцию можно остановить на стадии промежуточного альдимина, который легко гидролизуется до альдегида:
VII. Прочие производные карбоновых кислот
При ацилировании гидразина образуются гидразиды, при ацилировании гидроксиламина – гидроксамовые кислоты, а мочевина даѐт уреиды.
541
Некоторые гидразиды являются лекарственными препаратами (изониазид), гидроксамовые кислоты используются как реагенты для разделения и определения ионов металлов (дают красное окрашивание с солями железа и нерастворимые соли с солями меди), уреиды используются как лекарственные препараты (бромизовал, барбитураты и т.д.)
In vivo ацилирование происходит с помощью производного ацил-КоА, который, в свою очередь может образовываться из свободной карбоновой кислоты и коэнзима А с участием АТФ:
Большинство омыляемых липидов в организме образуется именно путѐм ацилирования спиртов с участием ацил-КоА. Структура коэнзима А (кофермента A) довольно сложна:
543
(кислотное омыление). При этом получаются глицерин и высшие карбоновые кислоты. Последние действием щелочи или соды переводят в мыла.
Моющая способность мыл связана с рядом сложных, вызываемых действием мыла, коллоидно-химических процессов. Главное заключается в том, что мыла являются ПАВами – они резко снижают поверхностное натяжение воды, вызывают смачивание частиц или поверхностей, обладающих водоотталкивающим действием, способствуют образованию устойчивой пены. Молекулы мыла, адсорбируясь на поверхности мельчайших капелек жиров или твердых частичек, загрязняющих тот или иной предмет или материал, удерживают их во взвешенном состоянии, т.е. образуют устойчивые эмульсии или суспензии. В виде последних жиры и грязь выводятся с поверхности и из пор ткани или др. материалов и предметов. Кроме того, мыла, являясь солями слабых кислот и сильных оснований, подвергаются гидролизу. Например:
C15H31COONa + HOH ↔ C15H31COOH + NaOH
Поэтому растворы мыл имеют щелочную реакцию, что также способствует эмульгированию жиров.
В жесткой воде моющая способность мыл резко снижается, растворимые натриевые или калиевые соли высших жирных кислот вступают в обменную реакцию с имеющимися в жесткой воде растворимыми кислыми карбонатами щелочноземельных металлов, главным образом кальция:
2C15H31COONa + Ca(HCO3)2 → (C15H31COO)2Ca + 2NaHCO3
Получающиеся при этом нерастворимые кальциевые соли высших жирных кислот образуют осадки.
Важнейшими представителями высших непредельных одноосновных кислот являются: олеиновая кислота C17H33COOH, линолевая
C17H31COOH и линоленовая C17H29COOH.
Олеиновая кислота в виде глицеринового эфира чрезвычайно распространена в природе. Ее строение выражается формулой:
CH3 – (CH2)7 – CH = CH – (CH2)7 – COOH
Олеиновая кислота – бесцветная маслянистая жидкость, легче воды, на холоде затвердевает в игольчатые кристаллы, плавящиеся при 14 . На воздухе она быстро окисляется и желтеет.
Молекула олеиновой кислоты способна присоединять два атома галогена:
CH3 – (CH2)7 – CH = CH – (CH2)7 – COOH + Br2 →
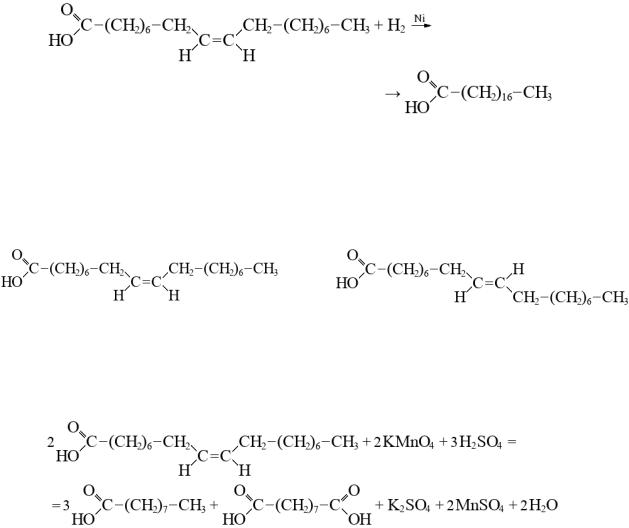
544
→ CH3 – (CH2)7 – CHBr – CHBr - (CH2)7COOH
9, 10 – дибромстеариновая кислота
В присутствии катализаторов, например Ni, олеиновая кислота присоединяет два атома водорода, переходя в стеариновую кислоту.
Олеиновая кислота представляет собой цис-изомер (все природные непредельные высокомолекулярные кислоты, как правило, относятся к цис- ряду). При действии на олеиновую кислоту оксидов азота она превращается в твердую элаидиновую кислоту, являющуюся транс-изомером:
|
NO, |
|
NO |
|
2 |
|
→ |
олеиновая кислота |
Элаидиновая кислота |
Жесткое окисление:
Линолевая C17H31COOH и линоленовая C17H29COOH кислоты еще более ненасыщены, чем олеиновая кислота. В виде сложных эфиров с глицерином – глицеридов – они являются главной составной частью льняного и конопляного масел. Их формулы:
CH3 – (CH2)4CH = CH – CH2 – CH = CH(CH2)7COOH
линолевая кислота
CH3 – CH2 – CH = CH – CH2 – CH = CH – CH2 – CH = CH(CH2)7COOH
линоленовая кислота
В молекуле линолевой кислоты две двойные связи. Она может присоединять четыре атома водорода или галогена. В молекуле линолевой кислоты три двойные связи, поэтому она присоединяет шесть атомов