

211
5)Твердость. Самый твердый – хром (режет стекло); самые мягкие – щелочные металлы – калий, натрий, рубидий и цезий – режутся ножом.
6)Плотность. Она тем меньше, чем меньше атомная масса металла и больше радиус атома. Самый легкий – литий (ρ=0,53 г/см3); самый тяжелый – осмий (ρ=22,6 г/см3). Металлы, имеющие плотность менее 5 г/см3 считаются
«легкими металлами».
7)Температуры плавления и кипения. Самый легкоплавкий металл – ртуть (т.пл. = -39°C), самый тугоплавкий металл – вольфрам (t°пл. = 3390°C). Металлы с t°пл. выше 1000°C считаются тугоплавкими, ниже – низкоплавкими.
Общие химические свойства металлов
Сильные восстановители: Me0 – nē → Men+
Ряд напряжений характеризует сравнительную активность металлов в окислительно-восстановительных реакциях в водных растворах.
1. Реакции металлов с неметаллами: а) кислородом: 2Mg + O2 = 2MgO
б) серой: Hg + S = HgS
в) галогенами: Ni + Cl2 –t°→ NiCl2
г) азотом: 3Ca + N2 –t°→ Ca3N2
д) фосфором: 3Ca + 2P –t°→ Ca3P2
е) водородом (реагируют только щелочные и щелочноземельные металлы): 2Li + H2 → 2LiH, Ca + H2 → CaH2
2. Реакции металлов с кислотами:
а) Металлы, стоящие в электрохимическом ряду напряжений до H восстанавливают кислоты-неокислители до водорода:
Mg + 2HCl → MgCl2 + H2.
2Al+ 6HCl → 2AlCl3 + 3H2.
6Na + 2H3PO4 → 2Na3PO4 + 3H2.
б) С кислотами-окислителями:
При взаимодействии азотной кислоты любой концентрации и концентрированной серной с металлами водород никогда не выделяется!
Zn + 2H2SO4(К) → ZnSO4 + SO2 + 2H2O,
4Zn + 5H2SO4(К) → 4ZnSO4 + H2S + 4H2O,
3Zn + 4H2SO4(К) → 3ZnSO4 + S + 4H2O,
2H2SO4(к) + Сu → Сu SO4 + SO2 + 2H2O, 10HNO3 + 4Mg → 4Mg(NO3)2 + NH4NO3 + 3H2O,
212
4HNO3(к) + Сu → Сu (NO3)2 + 2NO2 + 2H2O.
3. Взаимодействие металлов с водой:
а) Активные (щелочные и щелочноземельные металлы) образуют растворимое основание (щелочь) и водород:
2Na + 2H2O → 2NaOH + H2,
Ca+ 2H2O → Ca(OH)2 + H2.
б) Металлы средней активности окисляются водой при нагревании до оксида: Zn + H2O –t°→ ZnO + H2
в) Неактивные (Au, Ag, Pt) – не реагируют.
4. Вытеснение более активными металлами менее активных металлов из растворов их солей:
Cu + HgCl2 → Hg+ CuCl2,
Fe + CuSO4 → Cu+ FeSO4.
Металлы в свободном виде являются восстановителями. Однако реакционная способность некоторых металлов невелика из-за того, что они покрыты поверхностной оксидной пленкой, в разной степени устойчивой к действию таких химических реактивов, как вода, растворы кислот и щелочей.
Например, свинец всегда покрыт оксидной пленкой, для его перехода в раствор требуется не только воздействие реактива (например, разбавленной азотной кислоты), но и нагревание. Оксидная пленка на алюминии препятствует его реакции с водой, но под действием кислот и щелочей разрушается. Рыхлая оксидная пленка (ржавчина), образующаяся на поверхности железа во влажном воздухе, не мешает дальнейшему окислению железа.
Под действием концентрированных кислот на металлах образуется устойчивая оксидная пленка. Это явление называется пассивацией. Так, в
концентрированной серной кислоте пассивируются (и после этого не реагируют с кислотой) такие металлы, как Ве, Вi, Со, Fе, Мg и Nb, а в концентрированной азотной кислоте – металлы Аl, Ве, Вi, Со, Сг, Fе, Nb, Ni, РЬ, Тh и U.
При взаимодействии с окислителями в кислых растворах большинство металлов переходит в катионы, заряд которых определяется устойчивой степенью окисления данного элемента в соединениях (Nа+, Са2+,А13+,Fе2+ и
Fе3+).
Восстановительная активность металлов в кислом растворе передается рядом напряжений. Большинство металлов переводится в раствор соляной и разбавленной серной кислотами, но Сu, Аg и Нg – только серной (концентрированной) и азотной кислотами, а Рt и Аи – «царской водкой».

213
Закономерности изменения свойств оксидов и гидроксидов металлов в периодах и группах периодической системы отражены в таблице 2.5.1.2
Таблица 2.5.1.2 – Закономерности изменения свойств оксидов и гидроксидов металлов
В пределах малых периодов с ростом заряда ядра
Форма соединения |
|
Изменение свойств соединения |
|
|
|
|
|
|
|
|
|
Высший оксид и |
|
разность |
электроотрицательности |
элемента |
и |
соответствующий гидроксид |
кислорода (Э–) уменьшается |
|
|
||
|
|
основные свойства ослабевают |
|
|
|
|
|
кислотные свойства усиливаются |
|
|
|
|
|
окислительные свойства усиливаются |
|
|
|
|
|
восстановительные свойства ослабевают |
|
||
Летучее водородное |
|
разность электроотрицательности элемента-неметалла |
|||
соединение (образуют только |
и водорода (Э–) возрастает |
|
|
||
неметаллы) |
|
полярность связи возрастает |
|
|
|
кислотные свойства увеличиваются
Вглавных подгруппах с ростом заряда ядра
Высший оксид и |
|
основные свойства усиливаются |
|
соответствующий гидроксид |
|
кислотные свойства ослабевают |
|
Летучее водородное |
|
устойчивость ослабевает |
|
соединение (образуют только |
|||
|
кислотные свойства водного раствора увеличиваются |
||
неметаллы) |
|||
|
|
ЗАПОМНИ!!!
Свойства химических элементов, а также простых и сложных веществ, образуемых ими, закономерно изменяются в периодах слева направо и в главных подгруппах сверху вниз. По периоду слева направо уменьшаются: радиус атома, металлические свойства элементов и образованных ими простых веществ, основные свойства высших оксидов и соответствующих им гидроксидов; увеличиваются: радиус атома, неметаллические свойства элементов и образованных ими простых веществ, кислотные и окислительные свойства высших оксидов и соответствующих им гидроксидов, кислотные свойства водородных соединений неметаллов.
В главных подгруппах сверху вниз увеличиваются: радиус атома,
металлические свойства элементов и образованных ими простых веществ, основные свойства высших оксидов и соответствующих им гидроксидов, кислотные свойства водородных соединений неметаллов; ослабевают: неметаллические свойства элементов и образованных ими простых веществ, кислотные свойства высших оксидов и соответствующих им гидроксидов.
Природные соединения металлов. Минералы и горные породы,
содержащие металлы и их соединения и пригодные для промышленного получения металлов, называются рудами.
Отрасль промышленности, которая занимается получением металлов из руд, называется металлургией. Нахождение металлов в природе и их общие способы получения отражены в таблице 2.5.1.
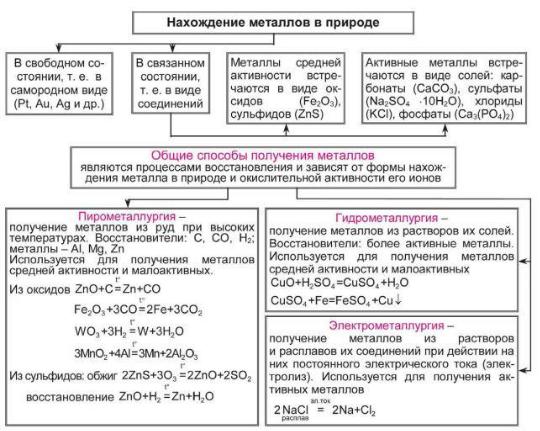
214
Рисунок 2.5.1 – Нахождение металлов в природе и их способы получения
Общие методы получения металлов и их очистки. Большинство металлов находится в природе в виде соединений с другими элементами (рисунок 2.5.1). Только немногие металлы встречаются в свободном состоянии, и тогда они называются самородными. Золото и платина встречаются почти исключительно в самородном виде, серебро и медь – отчасти в самородном виде; иногда попадаются также самородные ртуть, олово и некоторые другие металлы.
Добывание золота и платины производится или посредством механического отделения их от той породы. Все остальные металлы добываются химической переработкой их природных соединений.
Минералы и горные породы, содержащие соединения металлов и пригодные для получения этих металлов заводским путем, носят название руд. Главными рудами являются оксиды, сульфиды и карбонаты металлов.
Важнейший способ получения металлов из руд (пирометаллургический метод) (рисунок 2.5.1) основан на восстановлении их оксидов углем, оксидом углерода(II), водородом, алюминием.
Например, Cu2O + C = 2Cu + CO.
Подобным же образом производится выплавка чугуна их железных руд, получение олова из оловянного камня SnO2 и восстановление других металлов из оксидов.
Сернистые руды сначала переводят сернистые соединения в кислородные путем обжигания в особых печах, а затем уже восстанавливают полученные оксиды углем. Например, 2ZnS + 3O2 = 2ZnO + 2SO2; ZnO + C = Zn + CO.
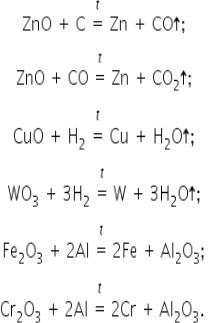
215
В тех случаях, когда руда представляет собой соль угольной кислоты, ее можно непосредственно восстанавливать углем, как и оксиды, так как при нагревании карбонаты распадаются на оксид металла и двуокись углерода.
Например, ZnCO3 = ZnO + CO2
Обычно руды, кроме химического соединения данного металла, содержат еще много примесей в виде песка, глины, известняка, которые очень трудно плавятся. Чтобы облегчить выплавку металла, к руде примешивают различные вещества, образующие с примесями легкоплавкие соединения – шлаки. Такие вещества называются флюсами. Если примесь состоит из известняка, то в качестве флюса употребляют песок, образующий с известняком силикат кальция. Наоборот, в случае большого количества песка флюсом служит известняк.
Во многих рудах количество примесей (пустой породы) так велико, что непосредственная выплавка металлов из этих руд является экономически невыгодной. Такие руды предварительно «обогащают», то есть удаляют из них часть примесей. Особенно широким распространением пользуется флотационный способ обогащения руд (флотация), основанный на различной смачиваемости чистой руды и пустой породы.
Для восстановления некоторых металлов из их оксидов применяют вместо угля водород, кремний, алюминий, магний и др.
Процесс восстановления металла из его оксида с помощью другого металла называется металлотермией. Если, в частности, в качестве восстановителя применяется алюминий, то процесс носит название алюминотермии.
Очень важным способом получения металлов является также электролиз
(электрометаллургический метод). Электролиз — это окислительно-
восстановительный процесс, протекающий под действием постоянного электрического тока, проходящего через раствор или расплав электролита.
216
Электролизом водного раствора можно выделить только те металлы, которые находятся в ряду активности правее алюминия (например, Zn, Ni, Sn, Cr, Pb, Co, Cu, Ag, Au, Pd и др.). Алюминий, магний, щелочные и щёлочноземельные металлы, титан извлекают электролизом расплавов их оксидов или хлоридов. Некоторые наиболее активные металлы получаются исключительно путем электролиза, так как все другие средства оказываются недостаточно энергичными для восстановления их ионов.
Гидрометаллургические методы. Гидрометаллургия – это выделение металлов из руд, концентратов и отходов производства с помощью водных растворов определённых веществ (химических реагентов).
Вначале металлы переводят в растворимые соединения. Затем их восстанавливают, используя либо химические реакции с сильными восстановителями, либо процессы вытеснения металлов из растворов их солей более активными металлами, либо электрохимическое восстановление из растворов.
Например, гидрометаллургическим методом извлекают медь из растворов её солей с помощью железа: CuSO4 + Fe = Cu↓ + FeSO4.
Серебро и золото восстанавливают из растворов солей этих металлов цинком. Покрытия из никеля и цинка получают электролизом растворов их солей.
Металлы используются для получения сплавой, которые нашли широкое применение (таблица 2.5.3) в промышленности.
Таблица 2.5.3 – Сплавы металлов и их применение
2.5.2 Значение и роль металлов
В отличие от элементов А-групп (C, H, O, N, P, K, Ca и др.) содержание металлов В-групп в живых организмах крайне невелико. Так, доля железа не
217
превышает 10−2%, а содержание Mn, Cu, Zn, Mo, V, Co, Ni, Cr составляет менее 10–3%. Поэтому их называют микроэлементами. Тем неменее соединения металлов В-групп выполняют ряд биологических функций в организмах.
Микроэлементы входят в состав биологически активных соединений (ферментов, гормонов, витаминов), которые регулируют обменные процессы веществ и клеточную энергетику, тканевое дыхание и другие жизненно важные процессы.
Среди элементов В-групп имеются также опасные для здоровья металлы, такие как Cd, Hg и др. Их соединения токсичны. Соединения никеля вызывают аллергию.
Физиологическое действие разных элементов зависит от их дозы. Поэтому при низкой концентрации соединение металла может действовать на организм как лекарство или выполнять жизненно нужные функции, тогда как при более высокой концентрации соединение этого же металла вызывает отравление. Например, важность для жизни цинка заключается в активизации этим элементом работы более 50 ферментов. Необходимая суточная норма поступления его в организм человека составляет 12–15 мг. Однако доза в 150 мг станет ядом для человека.
Области применения металлов: ядерная энергетика (U), производство осветительных приборов (Mo, W), медицина (протезы) (Ti, Ni, Au), легирующие добавки (W, Mo, Cr, V, Ni), ювелирные изделия (Au, Ag, Cu), защита от коррозии (Zn, Su, Ni, Cr), катализаторы (Pt, Fe, Ni, Pd), электротехника (Cu, Ag, Al) и др.
218
2.6 МЕТАЛЛЫ А – ГРУПП
ПЛАН
2.6.1Щелочные металлы.
2.6.2Бериллий. Магний. Щелочно-земельные металлы.
2.6.3Алюминий. Подгруппа галия.
2.6.4Металлы IVA группы.
2.6.1 Щелочные металлы
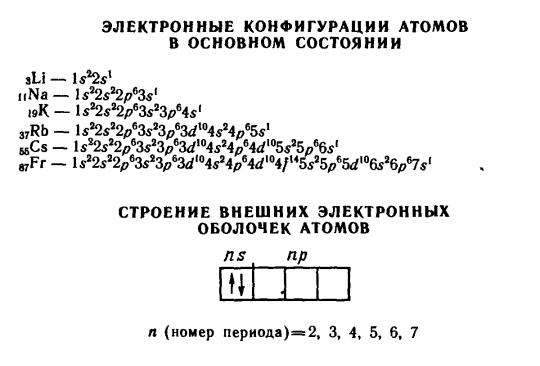
Щелочные металлы – элементы IА-группы (Li, Na, K, Rb, Cs, Fr) имеют на внешнем энергетическом уровне по одному s-электрону (s-элементы).
Они легко отдают валентные s-электроны, т.е являются сильными восстановителями. Щелочные металлы – типичные металлы, обладающие блеском, незначительной твёрдостью, малой плотностью и низкими температурами плавления, высокой электрической проводимостью, химически весьма активны, имеют малые значения энергии ионизации при относительно больших радиусах атомов или ионов. Как правило, они образуют соединения с ионным типом связи.
Все элементы IА-группы очень сходны по свойствам, что объясняется однотипным строением не только валентной оболочки, но и предвнешней (за исключения лития). С ростом радиуса атома в группе Li-Na-K-Rb-Cs-Fr ослабевает связь валентного электрона с ядром. Соответственно, в этом ряду энергия ионизации атомов щёлочных металлов уменьшается.
Все щелочные металлы имеют отрицательные стандартные окисслительно-восстановительные потенциалы большие по абсолютной величине, что характеризует их как сильные восстановители.
219
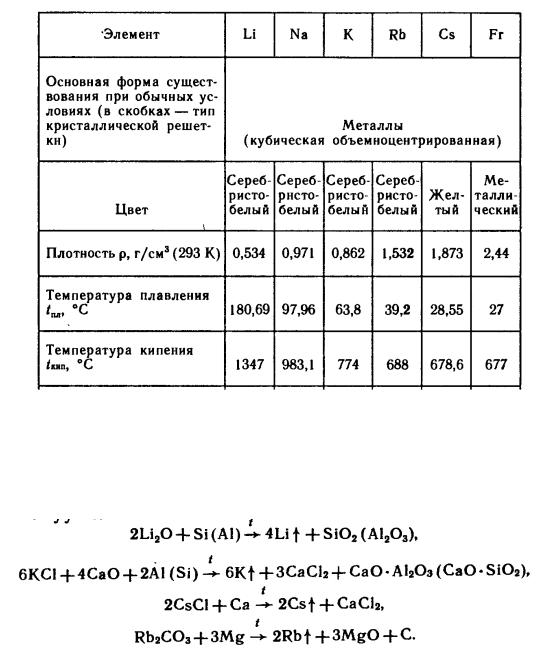
Проявляемые степени окисления: +1.
Физические свойства простых веществ представлены в таблице 2.6.1
Таблица 2.6.1 – Физические свойства щелочных металлов
Способы получения. Li, Na, K – электролиз расплавов их хлоридов в смеси с KCl, CaCl2 (калий, кальций) и NaCl (натрий) (Основной способ). Применяется также восстановление их оксидов, хлоридов, карбонатов алюминием, кремнием, кальцием, магнием при нагревании в вакууме:
Li и Na высокой частоты – элетролиз расплава смеси LiCl – LiBr и NaOH соответственно.
Химические свойства. Все щелочные металлы – очень сильные восстановители. Реакционная способность возрастает в ряду от Li к Cs. Щелочные металлы энергично реагирууют с разбаввленными растворами кислот. Комплексообразование для ионов щелочных металлов нехарактерно. Li
существенно отличается от остальныъх эелеметов группы. По роду свойств он ближе к Mg, чем к щелочным металлам.
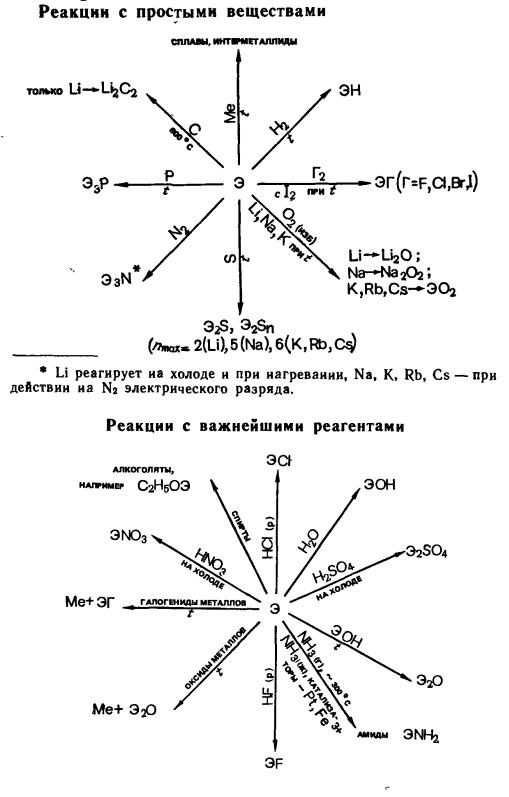
220
Соединения щелочных металлов с кислородом. Соединения щелочных металлов с кислородом: оксиды Э2О (ряд Li-Cs), пероксиды Э2О2 (ряд Li-Cs) и надпероксиды ЭО2 (ряд K-Cs) – кристаллические вещества с ионной решеткой, различной окраски (бесцветные – Li2O, Li2O2 , Na2O, желтые – Na2O2, К2O2, Rb2O, CsO2, оранжевые – Cs2O, KO2, розовый – К2O2, коричневый – RbO2); термически устойчивые. Пероксиды содержате диамагнитный ион O22-, надпероксиды – парамагнитный ион O2-
221
Некоторые характеристики кислородных соединений приведены ниже:
Способы получения. Э2О
Для Li: 4Li + O2 = 2Li2O,
Химические свойства.
1) Оксиды энергично реагируют с водой, образуя шелочи:
Э2О + Н2О → 2ЭОН
Интенсивность взаимодействия увеличивается в ряду Li2O-Cs2O, Э2О кроме Li2O, взаимодействуют с кислородом воздуха уже при комнатной температуре, образуя пероксиды: 2Э2О + О2 → 2Э2О2.
Пероксиды и надпероксиды взаимодействуют с H2O по схеме:

222
2Э2О2 + Н2О → 2ЭОН + Н2О2,
2ЭО2 + 2Н2О → 2ЭОН + Н2О2 + О2.
2) Аналогично протекает взаимодействие с кислотами:
2Э2О2 + Н2SO4 → 2Э2SO4 + Н2О2,
2ЭО2 + Н2SO4 → Э2SO4 + Н2О2 + О2.
3) Пероксиды и надпероксиды проявляют сильные окислительныесвойства:
Э2О2 + 2FeSO4 + 2Н2SO4 → Fe2(SO4)3 + Э2SO4 + 2Н2O
4) В реакциях с сильными окислителями они могут окисляться:
5Э2О2 + 2KMnO4 + 8Н2SO4 → 5О2 + 2MnSO4 + 5Э2SO4 + K2SO4 + 8Н2O
Соли щелочных металлов
Сульфиды Э2S – бесцветные кристаллические вещества с ионной решеткой.
Промышленный способ их получения – прокаливание сульфатов с углем:
4C + Э2SO4 →t 4CO + Э2S
В воде Э2S хорошо расторимы, частично гидролизуются:
Э2S + H2O ЭOH + ЭHS
Они являются сильными восстановителями:
Na2S + I2 → S + 2NaI.
Для многоосновных кислот известны средние и кислые соли щелочных металлов. Образование кислых солей — характерная особенность этих металлов. Склонность к образованию кислых солей и их термическая устойчивость увеличиваются от Li к Сs.
Почти все соли щелочных металлов хорошо растворимы в воде, являются сильными электролитами, малорастворимые— LiF, Li2СО3, Li3РО4, КСlO4,
К3[РtС16], Na[Sb(ОН) 6], РbСlO4, Rb2[РtС16], СsСlO4 и др.
Гидроксиды щелочных металлов
В ряду LiОН– СsОН наибольшее сходство наблюдается у гидроксидов
Nа, К, Rb и Сs.
Гидроксиды ЭОН – кристаллические белые вещества, сравнительно
223
легкоплавкие, термически очень устойчивые. При нагревании они испаряются без потери воды, только LiOН теряет воду, образуя Li2О.
ЭОН очень хорошо растворяются в воде, за исключением LiОН, хорошо в спиртах. Например, в метаноле СН3ОН, этаноле С2Н5ОН. В ряду LiOН— СsОН растворимость увеличивается.
Способы получения. LiOН, RbOН, СsОН — обменная реакция между растворами сульфатов щелочных металлов и гидроксидом бария:
Э2SО4 + Ba(OH)2 → ВаSО4 + 2ЭОН.
NaОН, КОН — 1) электролиз водных растворов хлоридов; 2) реакция каустификации:
Э2СО3 + Ca(OH)2 → СаСО3↓ + 2ЭОН.
Химические свойства. Гидроксиды щелочных металлов — сильные электролиты, в воде полностью диссоциированы на ионы. Они энергично поглощают из воздуха влагу (кроме LiОН) и СО2:
2ЭОН + СО2→Э2СО3 + Н2О.
При плавлении ЭОН разрушают стекло и фарфор:
2ЭОН + SiO2 → Э2SiO3 + Н2О,
при доступе кислорода — платину. Твердые гидроксиды и их концентрированные растворы разрушают живые ткани.
Применение и биологическая роль
Щелочные металлы и их соединения — важнейшие компоненты различных химических производств. Щелочные металлы широко используются для металлотерического получения ряда металлов, таких как Тi, Zг, Nb, Та, (Nа, К), в качестве добавок к некоторым сплавам, во многих органических синтезах (Li, Nа, К), для осушки органических растворителей (Nа).
Соединения щелочных металлов находят применение в мыловарении (Na2СОз), производстве стекла (Nа2СОз, К2СО3, Na2SO4, Li2OH), используются для отбелки и дезинфекции (Na2O2), в качестве удобрений (КС1, КNОз). Из NаС1 получают многие важные химические соединения (Nа2СОз, NаОН, С12).
Литий можно обнаружить в составе растительных и животных организмов, однако функции его – в жизнедеятельности неясны. Соли лития в больших концентрациях для человека опасны.
Натрий важен для большинства форм жизни, включая человека. В животных организмах ионы натрия вместе с ионами калия выполняют функции передатчиков нервного импульса. Ионы натрия играют также важную роль в поддержании водного режима организма. Соединения натрия нетоксичны.
Калий важен для всех живых существ. В растениях калий способствует

224
фотосинтезу и стимулирует процессы, связанные с прорастанием семян. В животных организмах калий необходим для нормальной работы мышечных клеток и нервной системы (вместе с ионами натрия). Однако в животных организмах ионов калия меньше, чем ионов натрия, и повышение концентрации калия оказывает вредное действие.
Биологическая роль Rb, Сs, Fг отсутствует. Цезий и его соединения нетоксичны. Токсичность рубия сравнительно мала. Франций токсичен из-за радиоактивности.
2.6.2 Бериллий. Магний. Щелочно-земельные металлы.
Элементы IIА-группы (Ве, Mg, Са, Sг, Ва, Rа) имеют на внешнем энергетическом уровне по два s-электрона (s-элементы).
Проявляемые степени окисления: +2.
Значения радиусов для атомов элементов IIА-группы в периодах, в сравнении со щелочными металлами, меньше, а энергия ионизации больше; в группах все аналогично. Обращает на себя внимание Ве, у него высокое значение Э.О., поэтому в его соединенияхпреимущественно ковалентные химические связи. У щелочноземельных – ионные.
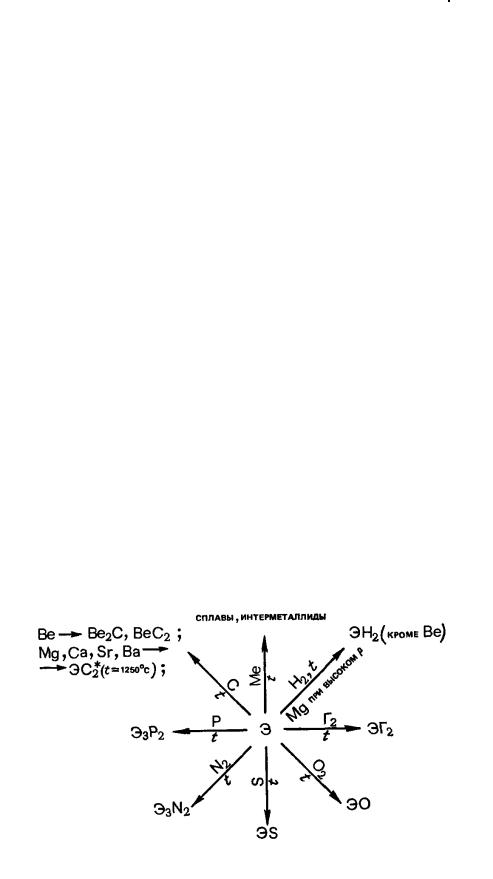
Физические свойства простых веществ представлены в таблице 2.6.2.1
Таблица 2.6.2.1 – Физические свойства щелочно-земельных металлов
Элемент |
Ве |
|
Мg |
Са |
Sr |
Ва |
Rа |
|
|
|
|
|
|
|
|
|
|
Основная форма (в |
|
Металл |
Металл (кубическая |
Металл (кубическая |
||||
скобках – тип |
|
|||||||
(гексагональная) |
гранецентрированная) |
объемноцентрированная) |
||||||
кристаллической |
||||||||
|
|
|
|
|
|
|
||
решетки) |
|
|
|
|
|
|
|
|
225
Цвет в компактном |
Свинцово- |
|
|
|
Сереб |
|
|
|
|
|
|
состоянии и в |
Серый |
|
|
ристо-белый |
Серебристо-белый |
Металлический |
|||||
порошкообразном |
|
|
|
|
|
|
|
|
|
блестящий |
|
сотоянии |
Темно-серый |
|
|
Серебристый |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
Плотность, г/см3 (293 К) |
1,8477 |
|
1,738 |
|
1,55 |
2,54 |
3,594 |
|
~5 |
||
|
|
|
|
|
|
|
|
|
|
||
Т пл. (°С) |
1287 |
|
649 |
|
839 |
769 |
729 |
|
700 |
||
|
|
|
|
|
|
|
|
|
|
||
Т кип. (°С) |
2970 |
|
1090 |
|
1484 |
1384 |
1637 |
|
1140 |
||
|
|
|
|
|
|
|
|
|
|
|
|
Способы получения. Ве, Мg, Са – электролиз расплавов их хлоридов в смеси с NаС1(Ве), КСl (Мg, Са) и СаF2(Са) (основной способ). Применяется также восстановление оксидов и фторидов металлов алюминием, магнием, углеродом, кремнием:
4ЭО + 2Аl = ЭО∙+ А12О3 + 3Э (Э = Са, Sr, Ва), при 700 °С,
ВеF2 + Мg = МgF2 + Ве, при 1000 °С,
МgО + C = СО + Мg, при 3000 °С,
2МgО + 2СаО + Si = 2СаO∙SiO2+ 2MgO, при 1200 °С.
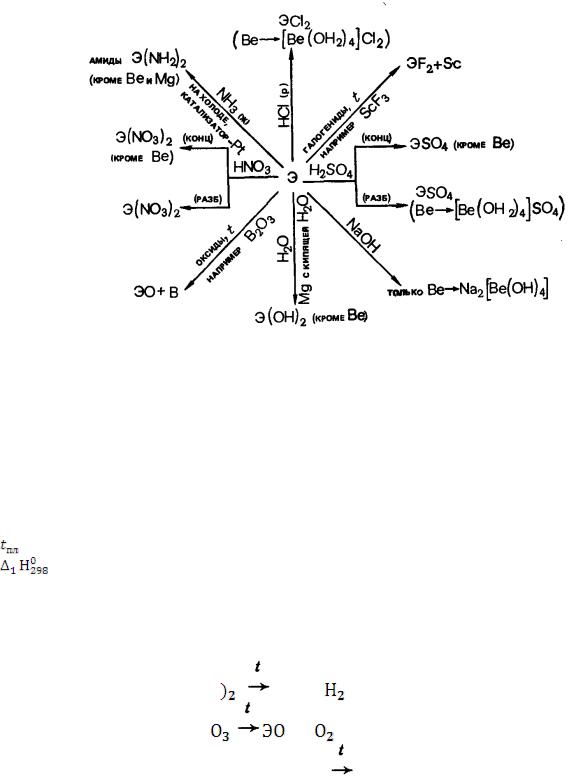
Химические свойства. Металлы группы ПА — сильные восстановители. Они довольно легко реагируют с большинством неметаллов; уже при обычных условиях интенсивно разлагают воду (кроме Ве и Мg); легко растворяются в кислотах; Ве реагирует и с кислотами, и со щелочами, образуя аква- и гидроксокомплексы.
Химическая активность повышается от Ве к Rа. По химическим свойствам Ве существенно отличается от остальных элементом группы. Mg так же во многих отношениях отличается от щелочноземельных металлов (Са-Ra).
Реакции с простыми веществами
226
Реакции со сложными веществами
Общая характеристика оксидов, гидроксидов, солей элементов IIА-
группы. Оксиды ЭО и пероксиды ЭО2 (ВеО2 не получен) — порошкообразные вещества белого цвета. Т пл. ЭО очень высоки и понижаются от Mg к Ва. Термическая диссоциация ЭО протекает очень трудно, теплоты образования (абсолютные значения) высокие. Термическая устойчивость ЭО2 повышается от МgО2 к ВаО2. В воде ЭО2 труднорастворимы.
От BeO к BaO основные свойства усиливаются. Некоторые характеристики ЭО приведены ниже:
Оксиды |
BeO |
MgO |
CaO |
SrO |
BaO |
,°С |
2580 |
2825 |
2630 |
2650 |
2020 |
*кДж/моль |
609 |
601,5 |
635,1 |
590,5 |
548 |
Способы получения. ЭО, кроме ВаО.
А) термическое разложение карбонатов или гидроксидов
элементов груп пы IIА: |
|
|
Э(OH |
ЭО + |
О (Э = Be, Mg), |
ЭС |
+С |
(Э = Be, Mg, Sr, Ca). |
ВаО — по реакции: 2Ba(NО3)2 |
2BaO + 4NО2 + О2. |
|
ЭО2 , кроме ВаО2 ,– нейтрализация гидроксидов Н2 О2 :
Э (ОН)2 + Н2О2 = ЭО2 + 2Н2О
ВаО2 — по реакции: 2ВаО + О2 = 2ВаО2.
Химические свойства. Оксиды Mg, Са, Sr, Ва – основного характера, ВеО проявляет амфотерные свойства.
227
Химическая активность ЭО увеличивается в ряду ВеО — ВаО. Оксиды, кроме ВеО, реагируют с водой; MgO реагирует частично с горячей водой: ЭО +  О = Э(ОН)2
О = Э(ОН)2
Оксиды легко реагируют с кислотами (ВеО при на гревании); ВеО реагирует также со щелочами:
ЭО+2НСl = ЭСl2+Н2О,
ВеО + 2НСl + 3H2О = [Be (О ]Сl2,
]Сl2,
ВеО + 2КОН + Н2 О = К2[Be (OН ],
],
BeO + 2KOH (тв.) = Ве
Ве + Н2О.
+ Н2О.
ВаО при нагревании в присутствии  образует ВаO2 :
образует ВаO2 :
2ВаO + O2 = 2ВаO3.
ЭO2 при растворении в воде подвергаются сильному гидролизу:
ЭO2+2 H2O = Э(ОН)2 + 
Они разлагаются кислотами, даже очень слабыми, например угольной: ЭО2 + СО2 = ЭСО3 + Н2 О2/
Пероксиды благодаря наличию иона [ O2]2 - проявляют окислительные свойст ва; при действии сильных окисли телей окисляются:
Ba + 2KI + 2Н2О =
+ 2KI + 2Н2О =  + Ba(OH
+ Ba(OH + 2KOH.
+ 2KOH.
Sr C
C =
=  + Sr
+ Sr C
C +Hg.
+Hg.
Пероксиды склонны к реакциям диспропорционирования:
2ВаO2 = 2ВаО +O2.
Соли. Сульфиды ЭS — кристаллические бес цветные вещества с ионной решеткой; термически устойчивы; малорастворимы в воде. Получают при взаимодействием простых веществ или прокаливанием сульфатов с углем: BaSО4 + 4C = BaS + 4CO.
В водном растворе с ульфиды сильно гидролизованы:
2ЭS + 2Н2О = Э(OH + Э(HS
+ Э(HS .
.
228
Вряду BaS– BeS гидролиз усиливается, BeS и MgS
гидролизуются полностью.
ЭS являются восстановителями: BaS+ =
=  .
.
Нитриды Э3 N2 — термически устойчивые кристалли ческие вещества. Получают их, как правило, нагреванием металлов в атмосфере N2: 3Э + N2 = Э3 N2.
Вводе нитриды необратимо гидролизуются:
Э3 N2 + 6H2O = 3Э(ОН + 2
+ 2
Многие соли оксокислот элементов группы IIA малораствори мы в воде. Это – сульфаты (кроме Ве и Mg), фосфаты, арсенаты, карбонаты, хроматы, оксалаты. С увели чением атомного номера металла растворимость солей и способность к образованию кристаллогидратов, как правило, уменьшаются.
При нагревании сульфаты, нитраты, карбонаты разлагаются по схемам:
2ЭSО4 = 2ЭO + 2SО2 + О2,
2Э = 2ЭO + 4
= 2ЭO + 4 + О2,
+ О2,
ЭСО3 = ЭО+ СО2.
Термическая устойчивость нитратов и карбонатов закономерно возрастает от соединений Ве к соединениям Ва.
При действии С на осадки карбонатов Са, Sr, Ва образуются растворимые гидрокарбонаты, которые при нагревании опять
на осадки карбонатов Са, Sr, Ва образуются растворимые гидрокарбонаты, которые при нагревании опять
переходят в карбонаты: ЭС |
+ С + Н2O = Э((НСО3 |
Гидроксиды Э(ОН |
– белые порошкообразные ве щества |
(кристаллическая решетка ионная). Они могут быть получены в безводном состоянии и в виде кристал логидратов с 1, 2, 3, 8 молекулами воды.
Растворимость в воде относительно невелика и увелич ивается при переходе от Ве( ОН к Ва(ОН
к Ва(ОН . Так же повышается термическая устойчивость и усиливаются основные свойства. Некоторые свойства гид роксидов элементов группы IIА приведены ниже:
. Так же повышается термическая устойчивость и усиливаются основные свойства. Некоторые свойства гид роксидов элементов группы IIА приведены ниже:
Гидроксид |
|
Be(ОН |
Mg(ОН |
Ca(ОН |
Sr(ОН |
Ba(ОН |
,°С |
|
Разлагаются до плавления |
|
535 |
408 |
|
кДж/моль |
906 |
924,7 |
985 |
965 |
941 |
|
Растворимость в воде |
Малорастворим |
0,00 |
0,16 |
0,81 |
3,89 |
|
при |
20°С, |
|
|
с |
|
|
безводного |
|
|
|
повышением |
|
|
вещества на |
100 г |
|
|
температуры |
|
|
H2O |
|
|
|
уменьшается |
|
|
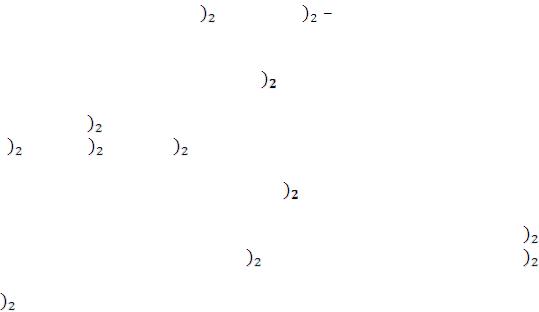
229
При нагревания Э(ОН , разлагаются, переходя в оксиды:
, разлагаются, переходя в оксиды:
Э(ОН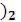 =
=  + H2O.
+ H2O.
Способы получения. Be(ОН , Mg(ОН |
обменные |
реакции |
||||
между растворимой солью металла и щелочью: |
|
|
||||
|
ЭСl2+2КОН = Э(ОН |
+ 2КСl. |
|
|
||
В случае Be(ОН |
следует избегать избытка щелочи. |
|
||||
Са (ОН , Sr(ОН |
, Ba(ОН |
– взаимодействие окси дов с водой: |
||||
|
ЭО + Н2О = Э(ОН . |
|
|
|||
Химические свойства. Характер диссоциации Э(ОН |
||||||
закономерно и зменяется |
от |
Be(ОН |
(амфотерный) до |
Ba(ОН |
||
(сильное основание). |
|
|
|
|
|
|
Be(ОН , растворяясь |
в кислотах и |
щелочах, образует |
аква - и |
|||
гидроксокомплексы: |
|
|
|
|
|
|
Be(ОН +2HCI + 2 Н2O = [ Be
+2HCI + 2 Н2O = [ Be ] Сl2,
] Сl2,
Be(ОН +2NaOH = Na2 [ Be
+2NaOH = Na2 [ Be ].
].
Mg(ОН проявляет только основные свойства, слабый электролит.
проявляет только основные свойства, слабый электролит.
Жесткость воды и ее устранение Жесткость воды – это совокупность свойств, обусловленных
содержанием в воде катионов кальция и магния. Анионами растворимых солей кальция и магния могут быть гидрокарбонат-ионы, сульфат-ионы и хлоридионы.
Различают временную (карбонатную) и постоянную жесткость. Временная жесткость обусловлена содержанием в воде
гидрокарбонатов кальция и магния. Временная жесткость легко устраняется кипячением:
Са(НСО3)2 = СаСО3 + СО2 + Н2О
Постоянная жесткость обусловлена наличием в воде сульфатов, хлоридов и других солей кальция и магния. Постоянную жесткость можно устранить, используя следующие способы.

230
а) Известково-содовый способ – к воде добавляют смесь гашеной извести и соды. При этом временная жесткость воды устраняется гашеной известью, а постоянная – содой:
Ca(HCO3)2 + Ca(OH)2 |
= 2CaCO3 |
+ 2H2O, |
CaCl2 + Na2CO3 |
= CaCO3 |
+ 2NaCl. |
б) Катионитный способ – воду пропускают через колонку, заполненную катионитом (катиониты – твердые вещества, содержащие в своем составе подвижные катионы, способные обмениваться на ионы внешней среды) Na2R, где R – анион. На катионите задерживаются ионы кальция и магния, а в раствор переходят ионы натрия, в результате чего жесткость воды уменьшается:
Na2R + Ca2+ = CaR + 2Na+,
Na2R + Mg2+ = MgR + 2Na+.
Через определенное время катионит необходимо регенерировать, т.е. восстановить первоначальные свойства. Для этого через катионообменные колонки пропускают насыщенный раствор поваренной соли, при этом происходят обратные процессы:
CaR + 2NaCl = Na2R + CaCl2,
MgR + 2NaCl = Na2R + MgCl2.
После такой промывки катионит снова можно использовать для умягчения воды.
Сумма временной и постоянной жесткости составляет общую жесткость воды, которая количественно выражается числом ммоль эквивалентов ионов кальция и магния, содержащихся в 1 л воды (таблица 2.6.2.2).
Таблица2.6.2.2 – Характеристика воды по степени жесткости
|
Характеристика воды по жесткости |
|
Общая жесткость, ммоль экв/л |
|
|
|
|
|
|
|
|
|
Очень мягкая вода |
|
Не более 1,5 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
Мягкая вода |
|
От 1,5 до 4 |
|
|
|
|
|
|
|
|
|
Вода средней жесткости |
|
От 4 |
до 8 |
|
|
|
|
|
|
|
|
Жесткая вода |
|
От 8 |
до 12 |
|
|
|
|
|
|
|
|
Очень жесткая вода |
|
Свыше 12 |
|
|
|
|
|
|
|
|
В жесткой воде плохо развариваются продукты, т.к. катионы кальция образуют нерастворимые соединения, взаимодействуя с белками пищи; плохо
231
завариваются чай, кофе; плохо мылится мыло из-за присутствия мало растворимых в воде стеаратов кальция и магния. Постоянное употребление жесткой воды может привести к отложению солей в организме человека.
Применение соединений магния и кальция и их роль в живой
природе |
|
|
|
|
|
|
Магний |
и |
кальций |
широко |
используются |
для |
|
металлотермического |
получения |
ряда |
металлов |
( Ti, |
U, |
|
редкоземельные элементы и др.). Бе риллиевые сплавы благодаря высокой химической и механической стойкости применяются в машиностроении, электронной и электротехнической промышленности; магниевые сплавы, как самые легкие, – в авиационной промышленности. Барий используется в высоковакуумной технике дл я поглощения остатков газов.
Соединения элементов группы IIА находят применение в радиоэлектронике (ВаТiOз – сегнетоэлектрик), строительстве (разнообразные соединения Mg и Са), для изготовления огнеупорных изделий (BeO, MgO), для осушки и очистки ряда веще ств (СаС , SrO, SrСОз) и в других областях. В настоящее врем я получены сверхпроводящие керамические материалы, содержащие ВаО и СаО.
, SrO, SrСОз) и в других областях. В настоящее врем я получены сверхпроводящие керамические материалы, содержащие ВаО и СаО.
Биологическая роль бериллия отсутствует. Бериллий и его соединения очень токсичны, берилий замещает магний в ферментах. Пары и пыль, содержащие соединения берилия , вызывают тяжелое заболевание легких.
Магний важен для всех живых существ. Глав ная его природная функция связана с процессом фото синтеза в растениях и микроорганизмах. Ионы магния принимают также участие в регулировании действия некото рых ферментов и клеточных систем. Соединения магния нетоксичны.
Кальций важен для всех форм жизни. Его биологи ческие функции разнообразны. Кальций входит в состав опорных и защитных частей организмов, его соединения образуют основу твердой части зубной ткани, скорлупы яйца. Ионы кальция содержатся в ряде белков, оказы вают существенное влияние на работу ферментных систем, на процессы свертывания крови, осмотическое равновесие в клетках. Соединения кальция нетоксичны.
Содержание стронция и бария в тканях животных и растений очень мало, и функции их соединений в организме не выяснены. Соли бария в значительных количествах про являют токсические свойства и действуют как сердеч ные яды. ВаСОз, несмотря на малую растворимость, опа сен для приема внутрь, так как он растворяется в соляной кислоте желудка.

232
2.6.3 Алюминий. Подгруппа галлия
Общая характеристика элементов. Элементы IIIA группы – Tl, In, Ga,
Al, B.
Электронные конфигурации атомов
5B – 1s22s22p1
13Al – 1s22s22p63s23p1
31Ga –1s22s22p63s23p63d104s24p1
49In – 1s22s22p63s23p63d104s24p64d105s25p1
81Tl – 1s22s22p63s23p63d104s24p64d104f145s25p65d106s26p1
Строение внешних электронных оболочек атомов
|
↑↓ |
↑ |
|
|
|
|
|
ns |
np |
|
|
|
|
|
n (номер периода) = 2,3,4,5,6 |
|
|
|||
Проявляемые степени окисления: |
|
|
|
|||
Элементы |
B |
Al |
Ga |
In |
Tl |
|
Степени окисления |
+3, –3 |
+3 |
+1, +2, +3 |
+1, +2, +3 |
+1, +3 |
|
элемента в соединениях |
||||||
|
|
|
|
|
||
ПРОСТЫЕ ВЕЩЕСТВА Основные физические свойства представлены в таблице 2.6.3.
Таблица 2.6.3 – Физические свойства элементов IIIA группы
Элемент |
B |
Al |
Ga |
|
In |
Tl |
|
|
|
|
|
|
|
Основная форма |
Неметалл, |
Металл |
|
|
|
|
существования (в |
Металл |
|
Металл |
Металл |
||
твердый |
(кубическая |
|
||||
скобках – тип |
(ромби- |
|
(тетра- |
(гексо- |
||
(ромбо- |
гранецентри- |
|
||||
кристаллической |
ческая) |
|
гональная) |
гональная) |
||
эдрическая) |
рованная) |
|
||||
решетки) |
|
|
|
|
||
|
|
|
|
|
|
|
Цвет |
Серый |
Серебристый |
|
Сере-бристо-белый |
||
кристаллический |
|
|||||
Темный |
— |
|
|
— |
|
|
аморфный |
|
|
|
|||
|
|
|
|
|
|
|
Плотность |
2,34 |
2,70 |
5,90 |
|
7,31 |
11,85 |
г/см3 (293 К) |
|
|||||
|
|
|
|
|
|
|
tпл. |
2300 |
660 |
29,78 |
|
156,17 |
303,5 |
tкип. |
3658 |
2467 |
2403 |
|
2080 |
1457 |
|
|
|
|
|
|
|
Способы получения. B – из приведенного сырья в несколько стадий:
Na2B4O7 10H2O → H3BO3 → B2O3 → B.
(природные бораты)
Al – электролиз глинозема (Al2O3) в расплавленном криолите AlF3 · 3NaF. Соединения Ga извлекаются из природных бокситов Al2O3 · xH2O,
233
In – из сернистых руд Zn, Pb и Cu;
Tl – из пирита FeS2 в виде растворов солей. Для выделения металлов в чистом виде растворы подвергаются гидролизу.
Химические свойства простых веществ. По проявляемым химическим свойствам Al, Ga, In и Tl относятся к металлам, бор — к типичным неметаллам.
В обычных условия, бор весьма инертен, но при высокой температуре активно реагирует с кислородом, галогенами, серой, азотом и углеродом. В кислотах, не являющихся окислителями, он не растворяется. Аморфный бор химически более активен, чем кристаллический.
Алюминий — довольно активный металл, однако прочная оксидная пленка на поверхности предохраняет его от окисления кислородом воздуха. С галогенами он реагирует при обычных условиях, с серой, азотом и углеродом
— при высокой температуре, растворяется в разбавленных кислотах и щелочах. Ga, In и Tl по химическим свойствам близки к алюминию, хотя щелочи на
Tl не действуют.
Реакции с простыми веществами
Реакции с важнейшими реагентами
234
2Ga + 3H2SO4 = Ga2(SO4)3 + 3H2,
2Al + 2NaOH + 6H2O = 2Na[Al(OH)4] + 3H2, 2B + 2KOH + 2H2O = 2KBO2 + 3H2 (аморфный B),
3Tl + 4HNO3 = 3TlNO3 + NO + 2H2O,
B + 3HNO3 = H3BO3 + 3NO2.
Общая характеристика оксидов, гидроксидов и солей элементов группы
Оксиды. Все элементы группы IIIA образуют оксиды типа Э2O3. Кроме того, таллий образует оксид Tl2O. Известны также оксиды и остальных элементов в более низких степенях окисления, но они практического значения не имеют.
Все оксиды — твердые вещества. B2O3, Al2O3, Ga2O3 — белого цвета, In2O3 — желтого цвета, Tl2O3 — темно-коричневого, Tl2O — гигроскопический порошок черного цвета.
B2O3 — кислотный оксид, растворимый в воде: B2O3 + 3H2O = 2H3BO3. Остальные оксиды Э2О3 в воде нерастворимы. Al2O3, Ga2O3, и In2O3
имеют амфотерный характер и растворяются в щелочах:
Э2О3 + 2NaOH + 3H2O = 2Na[Э(OH)4].
Tl2O3 имеет основный характер и в щелочах не растворяется. Оксиды Ga, In и Tl растворяются в кислотах с образованием солей:
Э2О3 + 6HNO3 = 2Э(NO3)3 + 3H2O.
Tl2O растворяется в воде с образование TlOH: Tl2O + H2O = 2TlOH
и в кислотах: Tl2O + 2HNO3 = TlNO3 + H2O.
Гидроксиды. Для всех элементов характерны гидроксиды типа Э(ОН)3, для таллия – также TlOH. Химические характер гидроксидов в ряду B(OH)3 – Tl(OH)3 изменяется довольно закономерно: H3BO3 – кислота, Al(OH)3, Ga(OH)3 и In(OH)3 – амфотерные основания с усиливающимися от Al к In основными свойствами, Tl(OH)3 имеет основный характер, TlOH – растворимое в воде сильное основание. Гидроксиды B, Al, Ga и In – белые вещества, Tl(OH)3 – красно-коричневого, TlOH – желтого цвета соединения.
Гидроксиды Al, Ga, In и Tl получаются осаждением щелочами из растворимых солей соответствующих металлов:
Э(NO3)3 + 3NaOH = Э(OH)3 + 3NaNO3.
235
Нерастворимые в воде гидроксиды Al, Ga, In и Tl при температурах около 100  легко теряют воду, переходя в оксиды: Э(ОН)3 → ЭООН → Э2О3
легко теряют воду, переходя в оксиды: Э(ОН)3 → ЭООН → Э2О3
Легко обезвоживается также TlOH: 2TlOH = Tl2O + H2O.
Вследствие амфотерности гидроксиды Al, Ga и In растворимы в щелочах: Э(ОН)3 + NaOH = Na[Э(ОН)4], с образованием соответственно алюминатов, галлатов и индатов. Такие соли устойчивы, если в их состав входят катионы активных металлов, но в водных растворах они сильно
гидролизованы.
Гидроксиды Al, Ga, In и Tl легко растворяются в кислотах с образованием солей катионов Al3+, Ga3+, In3+ и Tl3+. Первые три катиона бесцветны, Tl3+ имеет желтоватую окраску. Соли сильных кислот, в состав которых входят эти катионы, хорошо растворимы в воде, но сильно гидролизованы, чем обусловленная кислая реакция их растворов. Соли слабых кислот в водных растворах гидролизуют полностью:
Al2(CO3)3 + 6H2O = 2Al(OH)3 + 3H2O + 3CO2.
Катион Tl+ в водном растворе не гидролизует.
Соли. Все элементы рассматриваемой группы образуют с серой соединения типа Э2S3, таллий образует соединения Tl2S. Все сульфиды являются солями.
B2S3, Al2S3 и Ga2S3 полностью разлагаются водой:
Э2S3 + 6H2O = 2Э(OH)3 + 3H2S.
In2S3 и Tl2S3 не взаимодействуют не только с водой, но и с разбавленными кислотами.
Tl2S — труднорастворимая в воде соль, образуется при пропускании H2S через растворы солей трехвалентного таллия:
2TlCl3 + 3H2S = Tl2S + 2S + 6HCl.
Нитриды состава ЭN известны для B, Al, Ga и In. Все они — твердые кристаллические вещества. BN при обычных условиях химически инертен, остальные разлагаются щелочами:
2ЭN + 2NaOH + 6HOH = 2Na[Э(OH)4] + 2NH3.
BN получен в двух модификациях — графитоподобной (белый графит) и алмазоподобной (боразон). Особое значение имеет алмазоподобная форма BN, которая превосходит алмаз по твердости и термостойкости.
Элементы рассматриваемой подгруппы образуют с фосфором соединения типа ЭР. Все фосфиды устойчивы при обычных условиях, обладают полупроводниковыми свойствами. AlP, GaP и InP гидролизуют:
236
ЭР + 3HOH = Э(OH)3 + PH3.
Из карбидов наибольшее значение имеет карбид бора B4C – близкое по твердости к алмазу, хорошо проводящее электрический ток, химически инертное соединение.
С активными металлами образует соединения только бор, например, борид магния MgB2.
Борная кислота и бораты. Борная кислота – H3BO3 (ортоформа)
получается либо растворением в воде B2O3, либо вытеснением ее из буры Na2B4O7 действием сильных кислот:
Na2B4O7 + H2SO4 + 5H2O = 4H3BO3 + Na2SO4.
Борная кислота очень слабая, сравнительно малорастворима в воде. Особенность этой кислоты заключается в том, что при ее нейтрализации щелочами образуются соли не ортоформы, а различных неизвестных в свободном состоянии полиборных кислот, имеющих сложное строение, в
частности тетраборной кислоты H2B4O7:
2NaOH + 4H3BO3 = Na2B4O7 + 7H2O.
В избытке щелочи тетрабораты переходят в метабораты:
Na2B4O7 + 2NaOH = 4NaBO2 + H2O.
Применение и биологическая роль. Широкое применение находит
борная кислота и особенно бура. Последняя используется в медицине как дезинфицирующее средство, а также в стекольном, керамическом и других производствах. Бор и его соединения – нитрид BN, карбид B4C, фосфид BP – применяются как полупроводниковые, BN и B4C – сверхтвердые материалы.
Хорошее сочетание химических, механических и технологических свойств обусловливает широкое использование алюминия в технике, главным образом в виде различного рода сплавов. Область применения сплавов алюминия – от домашней утвари до современной авиатехники.
Такие соединения галлия, как GaP, GaAs, используются в качестве высокотемпературных полупроводниковых материалов. Широкое применение находят легкоплавкие сплавы на основе галлия в терморегуляторах и высокотемпературных термометрах.
Индий используется как добавка к подшипниковым сплавам. Сплавы, содержащие индий, применяются в качестве припоев для соединения металлов, стекла и керамики. InP, InAs — в полупроводниковой электронике.
Наибольшая часть добываемого таллия применяется в электронике, электротехнике и технике, использующей инфракрасное излучение, монокристаллы TlBr и TlI — для изготовления линз и призм приборах для
237
обнаружения теплового излучения, и приборов ночного видения, Tl2S — для изготовления фотоэлементов, чувствительных к инфракрасному излучению.
Бор – необходимый высшим растениям элемент.
Алюминий входит в состав тканей живых организмов и межклеточных растворов. Избыток алюминия в пище оказывает неблагоприятное воздействие на организмы.
Соединения таллия очень ядовиты.
2.6.4 Металлы IVA группы
Элементы IVA группы: C, Si, Ge, Sn, Pb.
Электронные конфигурации атомов в основном состоянии
6С – 1s22s22p2
14Si – 1s22s22p63s23p2
32Ge – 1s22s22p63s23p63d104s24p2
50Sn – 1s22s22p63s23p63d104s24p64d105s25p2
82Pb – 1s22s22p63s23p63d104s24p64d104f145s25p65d106s26p2
Строение внешних электронных оболочек атомов |
|
|||||||
|
|
ns |
|
np |
|
|
|
|
|
|
↑↓ |
↑ |
↑ |
|
|
|
|
|
|
|
|
|
|
|
|
|
n (номер периода) = 2, 3, 4, 5, 6. |
|
|
|
|
|
|
||
Элемент |
С |
Si |
Ge |
Sn |
Pb |
|||
Степени окисления |
-4, +2, +4 |
|
-4, +2, +4 |
+2, +4 |
+2, +4 |
+2, +4 |
||
элемента |
|
|||||||
|
|
|
|
|
|
|
|
|
Простые вещества Основные физические свойства представлены в таблице 2.6.4.
Таблица 2.6.4 – Физические свойства элементов IVA группы
Элемент |
|
С |
Si |
Ge |
|
Sn |
Pb |
|
|
|
|
|
|
||
Основная |
Неметалл с |
Неметалл с |
Элемент |
Металл 1. α- |
Металл |
||
форма |
атомной |
кристалличе |
атомной |
Модификация |
(кубическа |
||
существования |
кристаллическ |
ской |
кристалличе |
(кубическая |
я |
||
аллотропные |
ой решеткой |
решеткой |
ской |
типа алмаза). |
гранецентр |
||
модификаций |
1. |
Графит |
(кубическая |
решеткой |
2. Β- |
|
ированная) |
(в скобках – |
(гексагональн |
типа алмаза) |
(кубическая |
Модификация |
|
||
тип |
ая) |
|
типа алмаза) |
(тетрагональна |
|
||
кристаллическ |
2. |
Алмаз |
|
|
я) |
|
|
ой решетки) |
(кубическая) |
|
|
|
|
|
|
|
3. |
Карбин |
|
|
|
|
|
|
(гексагональн |
|
|
|
|
|
|
|
ая) |
|
|
|
|
|
|
Цвет |
1. |
Темно- |
Серебристо- |
Серовато- |
1. |
Серый |
Синевато- |
|
серого |
серый с |
белый |
2. |
Серебр |
серый |
|
|
2. |
Бесцветный |
металлическ |
|
исто-белый |
|
|
|
3. |
Черный |
им блеском |
|
|
|
|
|
порошок |
|
|
|
|
|
|
238
Плотность p, |
1. |
2,265 |
2,328 |
5,323 |
1. |
5,846 |
11,336 |
г/см3 (293 К) |
2. |
3,515 |
|
|
2. |
7,295 |
|
tпл., 0С |
|
3547 |
1410 |
937 |
|
231,7 |
327,4 |
tкип.,0С |
|
4827 |
2355 |
2830 |
|
2270 |
1740 |
|
(суб.) |
|
|
|
|
|
|
Способы получения. Углерод в форме древесного угля известен с древних времен. Он может быть получен при нагревании древесины без доступа воздуха, при обугливании животных остатков, неполном сгорании органических соединений (сажа). Графит и алмаз встречаются в природе, но в последнее время в основном их получают искусственным путём: графит – из смеси кокса и каменноугольной смолы, а алмаз – при очень высоком давлении и температуре из графита. Карбин получают синтетически при каталитическом окислении ацетилена и является наиболее стабильной формой углерода, а алмаз
–наименее стабильная форма.
В1990 г. из сажи, образованной при испарении графита в электрической дуге в атмосфере гелия, была выделена еще одна новая форма углерода, так называемые фуллерены. Это многогранники (своеобразные круглые молекулы), содержащие от 60 до 110 и более атомов углерода. Наиболее изученным является фуллерен С60, состоящий, как и футбольный мяч, из 13 пятиугольников и 20 шестиугольников.
Si – восстановление из SiO2 магнием:
t
SiO2 + Mg→Si + 2MgO
или углеродом в электрической печи:
t
SiO2 + 2C →Si + 2CO.
Высокой чистоты Si получают восстановлением SiCI4 цинком или водородом:
t
SiCI4 + 2Zn→Si + 2ZnCI2.
Ge, Sn и Pb – термическое восстановление их оксидных соединений с помощью H2, C, CO:
t
GeO2+2H2 → Ge+2H2O,
t SnO2+2C→ Sn+2CO,
t
PbO+CO → Pb+CO2.

239
Химические свойства. По химическим и физическим свойствам углерод и образуемые их соединения резко отличаются от остальных элементов группы.
Для Siи Ge соединений с подобными группировками не установлено. Sn и Pb образуют соединения, характерные для металлов.
При обычных условиях все аллотропные модификации углерода весьма инертны, другие элементы группы химически достаточно активны и взаимодействуют со многими веществами, как простыми, так и сложными. При увеличении температуры химическая активность всех веществ, образованных элементами группы, резко возрастает.
В соединения углерод и кремний проявляют степени окисления -4, +2, +4, Ge, Sn и Pb уменьшается.
Реакции с простыми веществами
С → карбиды; Si → силициды; Ge, Sn, Pb → сплавы
Реакции с важнейшими реагентами.
240
Si + 2NaOH+ H2O → Na2SiO3 + 2H2 ,
Sn + NaOH → Na2SnO2 + H2O ,
Ge + 4HNO3(конц) → GeO2 + 4NO2 + 2H2O , 3Pb + 8HNO3(разб) → 3Pb(NO3)2 + 2NO + 4H2O , Sn + 4H2SO4(конц) → Sn(SO4)2 + 2SO2 + 4H2O , Pb + 3H2SO4(конц) → Pb(HSO4)2 + SO2 + 2H2O .
Для всех элементов рассматриваемой группы известны водородные соединения типа ЭН4. Это – газы, устойчивость которых при переходе от СН4 к PbH4 уменьшается, а восстановительная способность увеличивается. Метан химически инертен, остальные газы имеют высокую реакционную способность, напримерSiH4самовоспламеняется на воздухе: SiH4+2O2 = SiO2 + 2H2O.
Вода разлагает SiH4, GeH4 и SnH4по схеме: ЭН4 + 2Н2О = ЭО2 + 4Н2. Метан содержится в природе, водородные соединения Si, Ge, Sn и Pb
образуются на ряду с водородом при разложении кислотами соединений или сплавов этих элементов с магнием: Mg2Si + 4HCI = SiH4 + 2MgCI2.
Углерод и кремний образуют галогениды только одного типа – ЭГ4 со всеми галогенами, Ge, Snи Pb – двух типов – ЭГ4 и ЭГ2 (кроме PbBr4 и PbI4).
Большинство галогенидов ЭГ4 – жидкости либо твёрдые вещества с молекулярной структурой. Солеобразный характер имеют только SnF4 и PbF4 . Галогениды углерода отличаются химической инертностью, в то время как группы галогениды остальных элементов активно взаимодействуют с водой:
nSiГ4+(2n+m)H2O → nSiO2∙mH2O+4nHГ.
Для галогенидов, особенно фторидов типа ЭГ4 , характерны реакции присоединения: ЭГ4+2НГ → Н2[ЭГ6].
По этой причине гидролиз SiF4протекает более сложно и выражается уравнением 3SiF4+2H2O→SiO2+2H2[SiF6].
Галогениды Ge, Sn и Pb типа ЭГ2 относятся к солям, причем GeГ2гидролизует практически полностью:
SnCl2 + H2O ↔ SnOHCI + HCI,
GeГ2 + H2O = Ge(OH)2 + 2HГ.
Для SnГ2 , и особенно для GeГ2 , характерны восстановительные свойства. Эти соли окисляются кислородом воздуха: 2ЭГ2 + О2 → ЭГ4 + ЭО2.
Оксиды. Для всех элементов рассматриваемой группы известны оксиды
241
типа ЭО и ЭО2. Оксиды углерода СО и СО2 – газы, все остальные – твердые, практически нерастворимые в воде вещества с преимущественно атомной структурой.
Наиболее сильно восстановительные свойства проявляются у СО при высокой температуре, у SiO, GeO и SnO – в щелочной среде:
SiO2 + 2NaOH = Na2SiO3 + H2.
PbO2 – сильнейший окислитель в кислой среде:
5PbO2 + 2MnSO4 + 3H2SO4 = 5PbSO4 + 2HMnO4 + 2H2O.
Гидроксиды и их производные
Углероду соответствует гидроксид H2CO3(угольная кислота). Н2СO3в свободном состоянии не получена. При растворении СО2 в воде устанавливается равновесие:
Н2О + СО2 ↔ Н2СО3 ↔ Н+ + НСО3- ↔ 2Н+ + СО32-.
Соли активных металлов угольной кислоты (карбонаты) устойчивы.
Для остальных элементов в зависимости от степени окисления состав гидроксидов можно выразить формулами Э(ОН)2 (кроме Si) и Э(ОН)4 . Это твердые, плохо растворимые в воде вещества. Так как соответствующие оксиды с водой не взаимодействуют, получают такие гидроксиды косвенным путём:
Pb(NO3)2 + 2NaOH → Pb(OH)2 + 2NaNO3,
SnCI4 + 4NaOH → Sn(OH)2 + 4NaCl.
Формулы гидроксидов Э(ОН)2 и Э(ОН)4 являются простейшими. В действительности осадки гидроксидов содержат переменные количества водый их состав выражается более общими формулами ЭО∙nН2О и ЭО2∙nН2О. Известна гексагидроксооловянная кислота состава H2[Sn(OH)6].
Как и у оксидов, у гидроксидов элементов степени окисления +2 восстановительные свойства наиболее выражены в щелочной среде, у гидроксидов четырехвалентных металлов – в кислой.
Амфотерные гидроксиды типа Э(ОН)2 и Э(ОН)4 растворяются в сильных щелочах и кислотах, образуя либо соли состава Na2[Э(ОН)4] (гидроксогерманиты, -станнистаннаты, -плюмбиты) и Na3[Э(OH)6] (гидроксогерманаты, -станнаты, -плюмбаты), либо соли катионов Э2+ и Э4+.
Соли кремниевой кислоты обычно представляют как производные несущей молекулы H2SiO3, например, Na2SiO3, CaSiO3 . Такие соли содержат полимерный анион, строение которого можно представить схемой:
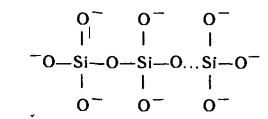
242
Применение и биологическая роль. Углерод наиболее широко применяется в металлургии как восстановитель в доменном процессе. В атомной энергетике и электротехнике используется графит.
Кремний находит применение в так называемых кремниевых приборах (радиоэлектроника, вычислительная техника, оптика, электротехника и т.д.). Карбид кремния SiC по твердости уступает только алмазу, в связи с чем используется как образцовый материал, а также как полупроводниковый. Широко применяется кварцевое стекло SiO2 . Кремний входит в состав многих сплавов железа и цветных металлов.
Германий используется в качестве полупроводникового материала. На основе GeO2 готовят специальные оптические стекла. Олово применяется для производства различных сплавов и белой жести для консервной промышленности. Свинец используется в свинцовых аккумуляторах, в производстве кабелей и химической промышленности в качестве защитного покрытия, в антифрикционных и типографических сплавах, а атомной энергетике и рентгенотехнике как поглотитель излучений. Оксид свинца используется при производстве красок и хрусталя.
Углерод – главный элемент всех органических соединений, известен на земле и в космосе. Благодаря углероду возникло все богатство и разнообразие видов растений и животных. На углеродной основе построена вся жизнь в биосфере. Кремний играет большую роль в биологической круговороте, никакой организм не может существовать без кремния. Избыток и недостаток кремния в организме болезненно сказывается на его развитии.
Германий и олово являются биологически активными элементами, однако их физические и биохимические свойства изучены пока недостаточно. Велика также биогенная роль свинца. Он входит в состав многих растений и животных (от тысячных до миллионных долей процента). Вместе с тем соединения свинца ядовиты и при длительном взаимодействии даже следовых количеств может привести к тяжелым заболеваниям, так как избыточный свинец накапливается в организме.
243
2.7 ПЕРЕХОДНЫЕ ЭЛЕМЕНТЫ (МЕТАЛЛЫ В – ГРУПП)
ПЛАН
2.7.1Общая характеристика d-элементов.
2.7.2Общая характеристика элементов групп меди и цинка.
2.7.3Общая характеристика элементов группы хрома
2.7.4Общая характеристика элементов группы марганца.
2.7.5Общая характеристика элементов семейства железа.
2.7.1 Общая характеристика d-элементов
К d-элементам относят те элементы, атомы которых содержат валентные электроны на (n – 1)d ns-уровнях и составляют побочные (IIIВ– VIIВ, IВ, IIВ) подгруппы. d-Элементы принято называть главными переходными элементами. Эти элементы занимают в периодической таблице переходное положение между электроположительными s-элементами и электроотрицательными p-элементами (таблица 2.7.1).
Таблица 2.7.1 – s, p, d элементы в таблице Менделеева
d-Элементы (все металлы) образуют три переходных ряда: 4– 6-ой. Строение атомов. Электронное строение атомов d-элементов
определяет их химические свойства. 3d-Элементы по химическим свойствам существенно отличаются от 4d- и 5d-элементов. При этом элементы IVВ– VIIВ подгрупп очень схожи по многим химическим свойствам. Это сходство обусловлено лантаноидным сжатием, которое из-за монотонного уменьшения радиусов при заполнении 4f-орбиталей приводит к практическому совпадению радиусов циркония и гафния, ниобия и тантала,
244
молибдена и вольфрама, технеция и рения. Элементы этих пар очень близки по физическим и особенно по химическим свойствам; их иногда называют элементами-близнецами.
Атомы d-элементов характеризуются общей электронной формулой (n – 1)d1–10ns0–2. Некоторые из тяжелых d-элементов не являются полными электронными аналогами. По мере увеличения числа d-электронов в периоде они могут переходить с одного уровня на другой для достижения требуемой правилами Гунда одной из наиболее устойчивых конфигураций (d5, d10). Такие переходы реализуются, например, в случае Cr(3d54s1), Cu(3d104s1), Mo(4d55s1), Ag(4d105s1). Обращает на себя внимание тот факт, что в одной подгруппе существуют элементы с разными электронными конфигурациями,
например, V(3d34s2), Nb(4d45s1) и Ta(5d36s2); Ni(3d84s2), Pd(4d105s0) и
Pt(5d96s1). Палладий является единственным d-элементом с незаполненным s- уровнем.
Проявление степени окисления. d-элементы проявляют только положительную степень окисления. Но в отличие от металлов IА и IIА групп для d-элементов валентными являются не только электроны внешнего слоя, расположенные на s-подуровне, но и неспаренные электроны d-подуровня предвнешнего слоя. Поэтому они, как и р-элементы, в соединениях могут проявлять несколько значений валентности или степени окисления (в зависимости от числа электронов, принимающих участие в образовании связей). Причем с увеличением номера группы от I до VII количество возможных значений степени окисления атомов d-элементов в соединениях возрастает. Величина же высшей степени окисления при этом совпадает с номером группы, в которой d-элементы находятся в таблице Д.И. Менделеева.
Например, ванадий находится в VВ группе. На внешнем слое его атома находятся 2s-электрона которые легко распариваются при возбуждении. За счет их будет проявлять степень окисления +2, но у ванадия валентными будут и неспаренные электроны, расположенные на 3d- подуровне. С учетом их может проявлять степень окисления +3, +4 и +5 (в зависимости от количестваd-электронов, участвующих вместе сsэлектронами в образовании связей).
Марганец находится в VIIB группе. У его атомов на внешнем слое находятся 2s-электрона. За счет их в возбужденном состоянии Mn может проявлять степень окисления +2. Кроме того, он может проявлять степень окисления +3, +4, +5, +6 и +7 (в зависимости от числа d-электронов, принимающих участие в образовании связей).
Элементы IВ группы, кроме степени окисления +1, могут проявлять и более высокие значения степени окисления: +2 (Cu) или +3 (Au).
Из элементов VIIIВ группы максимальную степень окисления +8 могут проявлять только Fe, Ru и Os. У остальных она ниже, чем +8.
С повышением степени окисления d-элементов основные свойства их соединений уменьшаются, а кислотные свойства, наоборот, возрастают.
245
Соединения (оксиды, гидроксиды), в которых d-элементы находятся в своей низшей степени окисления (+1 или +2), как правило, обладают основными свойствами. Если степень окисления d-элемента в соединении (оксиде, гидроксиде) равна +3 или +4, то ему присущи амфотерные свойства. И, наконец, соединения, в которых d-элементы проявляют свою высшую степень окисления (от +5 и выше), обладают кислотными свойствами.
Чем выше степень окисления d-элемента в соединении, тем в большей мере присущи ему окислительные свойства.
Изменение атомных радиусов, энергии ионизации и сродства к электрону в периодической системе. В целом в пределах каждого периода с ростом заряда ядра и накопления электронов на внешнем электронном уровне радиусы атомов убывают.
Наибольшее уменьшение радиусов наблюдается у элементов малых периодов, так как у них происходит заполнение внешнего электронного уровня. В больших же периодах в пределах семейств d- и f-элементов наблюдается более плавное уменьшение радиусов. Это уменьшение называется соответственно d- и f-сжатием.
Закономерные изменения свойств элементов четвертого периода (Fе, Со, Ni) проявляются значительно четче, чем у остальных элементов VIIIБгруппы. Поэтому Fе, Со, Ni объединены в семейство железа. Другие шесть элементов – в семейство платины.
Особенности химии d-элементов в сравнении с химией s- и p- элементов:
1.У d-элементов лишь небольшая часть валентных электронов делокализована по всему кристаллу (тогда как у щелочных и щелочноземельных металлов валентные электроны полностью отданы в коллективное пользование). Остальные d-электроны участвуют в образовании направленных ковалентных связей между соседними атомами. Таким образом, эти элементы в кристаллическом состоянии обладают не чисто металлической связью, а ковалентно-металлической. Поэтому все они твердые (кроме Hg) и тугоплавкие (за исключением Zn, Cd) металлы. Наиболее тугоплавки металлы VВ и VIВ подгрупп.
2.Вследствие незаполненности d-оболочек и наличия близких по энергии незаполненных ns- и np-уровней, d-элементы склонны к комплексообразованию; их комплексные соединения, как правило, окрашены
ипарамагнитны.
3.d-Элементы чаще, чем элементы главных подгрупп, образуют соединения переменного состава (оксиды, гидриды, карбиды, силициды, нитриды, бориды). Кроме того, они образуют сплавы между собой и с другими металлами, а также интерметаллические соединения.
4.Для d-элементов характерен большой набор валентных состояний и, как следствие этого, изменение кислотно-основных и окислительновосстановительных свойств в широких пределах.
Соединения, в которых d-элементы находятся в низшей степени
246
окисления, образуют кристаллы ионного типа, в химических реакциях проявляют основные свойства и являются, как правило, восстановителями.
Устойчивость соединений, в которых d-элементы находятся в высшей степени окисления (равной номеру группы), увеличивается в пределах каждого переходного ряда слева направо, достигая максимума для 3dэлементов у Mn, а во втором и третьем переходных рядах – у Ru и Os соответственно. В пределах одной подгруппы стабильность соединений высшей степени окисления уменьшается в ряду 5d > 4d > 3d.
Соединения, в которых d-электроны находятся в промежуточных степенях окисления, проявляют амфотерные свойства и окислительновосстановительную двойственность.
5.Сходство d-элементов с элементами главных подгрупп в полной мере проявляется у элементов третьей группы ns 2np 1 и (n – 1)d 1ns 2. С возрастанием номера группы оно уменьшается; элементы VIIIА подгруппы – газы, VIIIВ – металлы. В первой группе снова появляется отдаленное сходство (все элементы – металлы), а элементы IВ подгруппы – хорошие проводники; это сходство усиливается во второй группе, так как d-элементы Zn, Cd и Hg не участвуют в образовании химической связи.
6.d-элементы IIIВ–VIIВ подгрупп в высших степенях окисления по свойствам подобны соответствующим p-элементам. Так, в высших степенях окисления Mn (VII) и Cl (VII) являются электронными аналогами. Подобие электронных конфигураций (s 2p 6) приводит к подобию свойств соединений
семивалентных марганца и хлора. Mn2O7 и Cl2O7 в обычных условиях малоустойчивые жидкости, являющиеся ангидридами сильных кислот с общей формулой НЭО4. В низших степенях окисления марганец и хлор имеют различное электронное строение, что обусловливает резкое отличие свойств их соединений. Например, низший оксид хлора Cl2O – газообразное вещество, являющееся ангидридом хлорноватистой кислоты (HClO), тогда как низший оксид марганца MnO представляет собой твердое кристаллическое вещество основного характера.
7.Как известно, восстановительная способность металла определяется не только его энергией ионизации (М – ne – → Мn +; +∆H иониз), но и энтальпией гидратации образовавшегося катиона (М n + + mH2O → М n +•mH2O; –∆H гидр). Энергии ионизации d-элементов в сравнении с другими металлами велики, но они компенсируются большими энтальпиями гидратации их ионов. Вследствие этого электродные потенциалы большинства d-элементов отрицательны.
В периоде с ростом Z восстановительные свойства металлов уменьшаются, достигая минимума у элементов IВ группы. Тяжелые металлы VIIIВ и IВ групп за свою инертность названы благородными.
Окислительно-восстановительные тенденции соединений d-элементов определяются изменением устойчивости высших и низших степеней окисления в зависимости от положения их в периодической системе. Соединения с максимальной степенью окисления элемента проявляют
247
исключительно окислительные свойства, а с низшей – восстановительные. Поскольку для d-элементов в пределах подгруппы устойчивость
высших степеней окисления сверху вниз растет, то окислительные свойства соединений высшей степени окисления резко падают.
8. На кислотно-основные свойства гидроксидов d-элементов влияют те же факторы (величина ионного радиуса и заряд иона), что и на гидроксиды p- элементов.
Гидроксиды низших степеней окисления d-элементов обычно проявляют основные свойства, а отвечающие высшим степеням окисления – кислотные. В промежуточных степенях окисления гидроксиды амфотерны.
В пределах одной подгруппы гидроксиды d-элементов одинаковой степени окисления характеризуются увеличением основных свойств при движении сверху вниз.
Способность к комплексообразованию. d-элементы, как правило,
являются типичными комплексообразователями, причем максимальная способность к комплексообразованию принадлежит элементам VIIIБ-группы. Многие соединения d-элементов имеют характерную окраску и проявляют (наряду с простыми веществами) каталитическую активность. Для d- элементов известны почти все типы комплексных соединений.
Свойства простых веществ. Простые вещества, которые образуют d- элементы, обладают более высокими температурами плавления и имеют большую плотность, чем металлы, образованные s-элементами. Это объясняется тем, что в образовании металлической связи у d-элементов принимают участие не только электроны внешнего слоя (один или два), но и неспаренные электроны с d-подуровня предвнешнего слоя. В результате металлическая связь становится более прочной.
Металлы, образованные d-элементами, являются лучшими проводниками электрического тока, чем щелочные и щелочноземельные металлы. Особенно это характерно для тех металлов, атомы которых имеют только один внешнийs-электрон и полузаполненный (Cr, Mo) или заполненный (Cu, Ag, Au) d-подуровень предвнешнего слоя.
Роль в биологических системах. Из всех d-элементов наибольшую биологическую активность имеют элементы четвертого периода: Те, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, а также некоторые d-элементы пятого и шестого периодов.
Mn, Fe, Co, Ni, Cu– эссенциальными (жизненнонеобходимыми) элементами, необходимыми для всех известных форм жизни на Земле.
Каждый из этих элементов имеет свою функцию. Так, марганец ускоряет созревание лейкоцитов (эритроцитов). Кобальт предотвращает новообразованию ретикулоцитов и утилизации резервного железа. Никель усиливает действие кобальта. Медь обеспечивает всасывание железа в желудке и участвует в синтезе гемоглобина. Такая высокая организация процесса кроветворения доказывает выборочное распределение функций микроэлементов и их высокоэффективное использование.

248
В большинстве биогенных d-элементов достаточно полно изучены. Нарушение количественного состава химических элементов играют важную роль в возникновении некоторых заболеваний. Так, доказано, что частота возникновения сердечно-сосудистых заболеваний и сахарного диабета (частота заболевания) повязанная каким-то образом с содержанием d- элементов (хром, медь, цинк и кадмий). Некоторые эндемичные заболевания связаны с избытком или недостатком таких d-элементов: кобальта, цинка, меди, марганца и т.д..
2.7.2 Общая характеристика элементов групп меди и цинка
IВ ГРУППА. К d-элементам IВ группы относятся металлы медь, серебро, золото.
Эти металлы известны человечеству с глубокой древности, так как встречаются в природе в самородном виде. Медь относится к достаточно распространенным элементам, серебро и особенно золото – к редким элементам. Медь и серебро встречаются в природе преимущественно в виде соединений с серой: Cu2S (халькозин, или медный блеск), CuS (ювелин), CuFeS2 (халькопирит, или медный колчедан), Ag2S (аргентит).
Степени окисления: (медь, серебро, золото): +1, +2, +3. Наиболее устойчивой для меди является С.О. +2, для серебра +1, золота +3.
Медь, серебро, золото находятся в одной группе со щелочными металлами, но в отличие от них d-металлы имеют малый атомный радиус. Меньшие по размеру атомы более плотно располагаются в решетке, вследствие чего силы притяжения между ними велики. Этим объясняется
высокая плотность и высокая температура плавления металлов подгруппы меди.
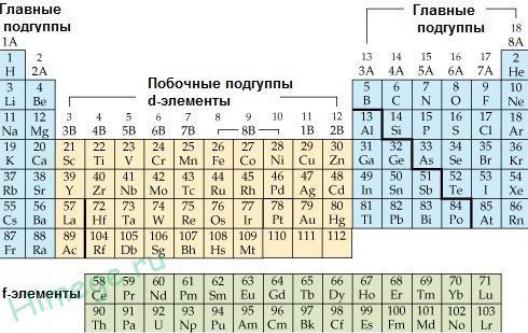
Химические свойства. Химическая активность Сu, Ag, Аu невелика и в ряду Сu–Аg уменьшается. Медь, серебро и золото из простых веществ легче всего реагируют с галогенами. Из растворов кислот и воды водород они не вытесняют. Исключение составляет взаимодействие Сu с конц. НСl и Ag с
249
конц. НI. Сu и Ag легко растворяются в кислотах, содержащих анионокислитель. Особенно устойчивым к действию кислот является золото. Оно растворяется только в «царской водке» (смеси конц. кислот азотной и соляной): Au + HNO3 + 4HCl = H[AuCl4] + NO↑ + 2H2O.
По отношению к щелочам в отсутствие окислителей данные вещества устойчивы. Для них характерно комплексообразование. Сu и Ag обладают высокой каталитической активностью.
Реакции со сложными веществами
250
Ag + 2HNO3(конц) t= AgN03+NO2 + H2O,
Cu + 2H2SO4(конц) t= CuSO4 + SO2 + 2H2O,
Cu + 4HNO3 (конц) = Cu(NO3)2 + 2NO2 + 2H2O, 2Au + 3Cl2+2HCl = 2H [AuCl4].
Cu + Cl2 = CuCl2.
2Cu + O2 = 2CuO (черный);
4Cu + O2 = 2Cu2O (красный).
В сухом воздухе медь не изменяется, так как образуется защитная оксидная пленка. Но в присутствии влаги и диоксида углерода поверхность меди покрывается зеленоватым налетом основного карбоната меди (патина)
(CuOH)2CO3: 2Cu + О2 + СО2 + H2O = (CuOH)2CO3
Соединения меди (I) и (II)
Оксид меди (Ι) не реагирует с водой, имеет основной характер, взаимодействует с кислотами с образованием солей:
Cu2O + 2HCl = 2CuCl + H2O
Гидроксид меди (Ι) CuOН – не существует.
Соли меди (Ι). Все средние соли меди (Ι) или не растворимы в воде (CuCl, Cu2S), или неустойчивы в водном растворе (Cu2SO4). Например, сульфат меди (Ι) диспропорционирует, образуя сульфат меди (ΙΙ) и свободную медь:
Cu2SO4 = CuSO4 + Cu.
Однако катион меди (Ι) образует устойчивые комплексные соли с аммиаком, тиосульфат-ионом, тиоцианат-ионом, цианид-ионом:
Cu2O + 4NH3 + H2O = 2Cu[(NH3)2]OH,
CuI + 2Na2S2O3 = Na3[Cu(S2O3)2] + NaI,
CuCl + 2КCN = К[Cu(CN)2] + КCl.
Оксид и гидроксид меди (ΙΙ). CuO и соответствующий ему гидроксид Cu(OН)2 обладают амфотерными свойствами с преобладанием основных свойств.
251
СuО+2НСl (конц.) = СuСl2+Н2O,
СuО+4НСl (конц., изб) = Н2 [СuСl4]+Н2O,
СuO+2NаOН (конц) + Н2O = Nа2[Сu(ОН)4], CuO+2NaOH сплавление= Nа2СuO2+Н2O.
Проявляя основные свойства, они легко реагируют с большинством кислот, образуя соли меди (ΙΙ):
CuO + 2НBr = CuBr2 + H2O,
Cu(OН)2 + H2SO4 = CuSO4 + 2H2O.
Кислотные свойства проявляются при взаимодействии с концентрированными растворами щелочей. Например, с раствором гидроксида калия образуется тетрагидроксокупрат (ΙΙ) калия:
Cu(OН)2 + 2KOH = K2[Cu(OH)4].
Оксид меди (ΙΙ) не реагирует с водой и его гидроксид получают косвенно, действуя на растворимые соли меди (ΙΙ) разбавленными растворами щелочей.
Гидроксид меди (ΙΙ) осаждается в виде голубой студенистой массы:
CuSO4 + 2NaOH = Cu(OН)2↓ + Na2SO4.
При слабом нагревании гидроксид меди (ΙΙ) разлагается, превращаясь в черный оксид меди: Cu(OН)2 = CuО + H2O.
Гидроксид меди (ΙΙ) – слабое основание, поэтому растворимые соли меди (ΙΙ) в водном растворе подвергаются гидролизу и имеют кислую реакцию среды: 2CuSO4 + 2H2O ↔ (CuOН)2SO4 + H2SO4 или в ионной форме: 2Cu2+ + 2H2O ↔ 2CuOН+ + 2H+
Важнейшие соли меди (ΙΙ). Сульфат меди (ΙΙ) безводный представляет собой порошок белого цвета, который при поглощении воды синеет. Водный раствор его имеет сине-голубой цвет. Такая окраска обусловлена гидратированными ионами меди [Cu(H2O)4] 2+. Все растворимые соли меди (ΙΙ) кристаллизуются из водных растворов в форме кристаллогидратов, например, пентагидрат сульфата меди CuSO4∙5H2O (прозрачные синие кристаллы), гексагидрат нитрата меди Cu(NO3)2∙6H2O (синие кристаллы), дигидрат хлорида меди CuCl2∙2H2O (темно-зеленые кристаллы).
Соединения меди (ΙΙ) обладают умеренными окислительными свойствами. В водном растворе под действием достаточно сильных
252
восстановителей медь (ΙΙ) восстанавливается до меди (Ι). Например, с раствором йодида калия образуются йодид меди (Ι) и свободный йод:
2CuSO4 + 4KI = 2CuI + I2 + 2K2SO4.
Комплексные соли меди (ΙΙ). Катион меди (ΙΙ) образует большое число комплексных соединений (хелатные комплексы), в том числе с многоатомными спиртами, аминокислотами, этилендиамином и т.д., в которых чаще всего проявляет КЧ = 4.
CuSO4 + 4NH3 = Cu[(NH3)4]SO4,
СuСl + 2NН3 = [Сu (NH3)2]Cl, CuCl2 + 4NH3 = [Cu (NH3)4]Cl2,
СuO + 4NН3 + Н2O = [Сu(NН3)]4(ОН)2,
Сu(ОН)2 + 4NН3 = [Сu (NH3)4](ОН)2.
Среди катионных комплексов Сu аммиакаты типа [Сu(NН3)2]+ (бесцветный) и [Сu(NН3)4]2+ (темно-синий) особо устойчивы. Аквакомплексы Сu (I) малостойки, и кристаллогидраты для солей Си (I) нехарактерны. Большинство солей Сu (I) нерастворимы в воде, но легко растворяются в присутствии NН3. Для Сu (II), напротив, очень характерны аквакомплексы голубого цвета типа [Сu(ОН2)4]2+, что обусловливает голубую или синюю окраску водных растворов её, солей и большинства кристаллогидратов.
Безводные соли Сu (II) чаще всего бесцветны или окрашены в темнокоричневый либо черный цвет. В водном растворе они частично гидролизуются с образованием малорастворимых основных солей.
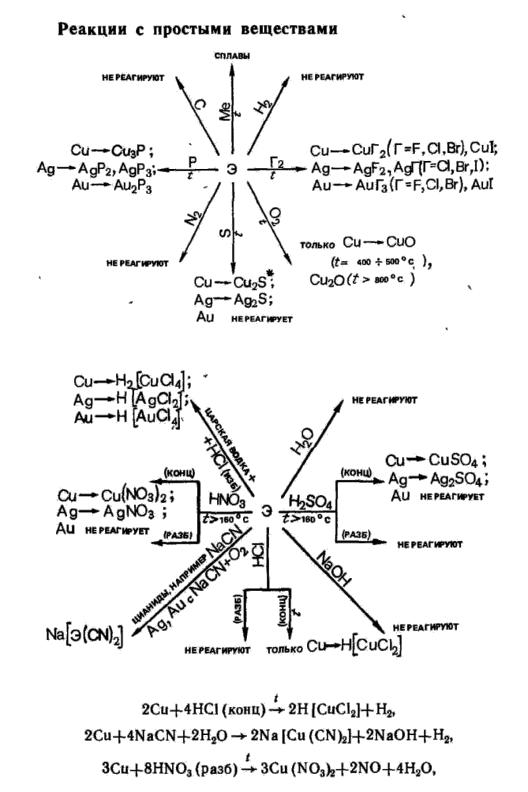
IIВ ГРУППА. К d-элементам IIВ группы относятся металлы цинк, кадмий и ртуть.
Металлы подгруппы цинка встречаются преимущественно в виде сульфидных руд: ZnS (вюрцит и сфалерит), CdS (гриконит), HgS (киноварь). Ртуть изредка встречается в самородном виде, вкрапленная в горные породы.
Каждый из этих элементов в своем периоде является последним элементом d-семейства, поэтому d-подуровень предвнешнего уровня полностью заполнен и устойчив. Поэтому в образовании химических связей могут участвовать только два s-электрона внешнего уровня.

253
Степени окисления. В соединениях цинк и кадмий имеют степень окисления +2 (ZnO, CdS), а ртуть имеет степени окисления, равные +1 и +2
(Hg+2Cl2, Hg2+1Cl).
Степень окисления ртути +1 объясняется образованием кластерного иона [– Hg–Hg – ]+2 (сокращенно Hg2+2) по донорно-акцепторному механизму: Hg0↓↑ + Hg+2 → [Hg↓↑Hg]+2.
Атом ртути Hg0, как видно, является донором двух 6s-электронов, а ион ртути Hg+2 – акцептором электронов за счет вакантной 6s-орбитали. Обобществление электронов приводит к образованию между атомами ртути σ-связи. В ионе [– Hg–Hg –]+2 ртуть двухвалентна, степень окисления +1.
Цинк, кадмий и ртуть сильно отличаются по свойствам от элементов главной подгруппы (бериллия, магния, кальция, бария). Восстановительные свойства их выражены значительно слабее, чем у элементов главной подгруппы. Это объясняется меньшими размерами атомов и, соответственно, более высокими значениями энергии ионизации элементов.
Закономерности изменения основных параметров такие же, как и для d-элементов других групп, а именно:
•радиусы атомов и ионов в подгруппе цинка сверху вниз увеличиваются, но незначительно и у кадмия и ртути радиусы атомов почти одинаковые;
•значение энергии ионизации сверху вниз в подгруппе увеличивается;
•химическая активность металлов от цинка к ртути уменьшается;
•восстановительные свойства убывают от цинка к ртути, т.к. стандартный электродный потенциал (Е0) возрастает от цинка к ртути. Химические свойства. Химическая активность в ряду Zn — Hg понижается. Zn и Cd и их соединения сходны. Растворяется Hg только в кислотахокислителях. Zn и Cd вытесняют водород из растворов кислотнеокислителей. При нагревании Zn, Cd, Hg реагируют с активными неметаллами. Элементы способны к комплексообразованию. Характерные
координационные числа: 4 — для Zn; 6 — для Cd; для Hg ( I I ) — 2 , 4 , 6
254
Реакции с простыми веществами
СПЛАВЫ, ИНТЕРМЕТАЛЛИДЫ
Реакции со сложными веществами
Zn + H2SО4 (разб) = ZnS04 + H2,
Zn + 2H2SO4 (конц) = ZnS04 + SO2 +2H2O, 4Zn+ 10HNO3(paзб) = 4Zn(NO3)2 + NH4NO3+3H2O,
t>1600C
Hg + 2H2S04 (конц) = HgSО4 + SО2 + 2H2O, 4Zn + 5H2SO4 = 4ZnSO4 + H2S + 4Н2O,
255
3Hg + 8HNO3 (разб) = 3Hg (NO3)2 + 2NO + 4Н2О,
6Hg + 8HNO3 (разб) = 3Hg2( N03)2 + 2NO + 4H2O,
Hg + 4HNO3(конц)=Hg(NO3)2 + 2NO2 + 2H2O,
t
3Hg + 6HCI+2HNО3 = 3HgCI2+2NO + 4H2O,
t
Zn + 2NaOH + 2H2O = Na2[Zn (OH)4]+ H2.
Образующаяся пленка защищает цинк от дальнейшего окисления. При обычных условиях цинк не взаимодействует с водой, так как образующаяся на поверхности пленка гидроксида цинка практически не растворима в воде.
2Zn + O2 + 2H2O = 2Zn(OH)2,
2Zn + O2 + CO2 + H2O = Zn2CO3(OH)2
При сжигании в воздухе образуется оксид цинка: 2Zn + O2 → 2ZnO.
Цинк окисляется хлором, бромом, йодом в водном растворе при комнатной температуре с образованием галогенидов цинка:
Zn + Cl2 = ZnCl2,
Zn + Br2 = ZnBr2,
Zn + I2 = ZnI2.
Соединения цинка Оксид цинка не реагирует с водой, имеет амфотерный характер,
взаимодействует с кислотами и щелочами с образованием солей:
ZnO + 2HCl = ZnCl2 + Н2O,
ZnO + 2NaOH + Н2O = Na2[Zn(OH)4],
восстанавливаются при действии сильных восстановителей:
ZnO + C = Zn + CO.
Гидроксид цинка. Zn(OH)2 – термически нестабильные соединения. разлагается при 125°С: Zn(OH)2 = ZnO + Н2О
Получают гидроксиды цинка и кадмия подщелачиванием растворов их солей.
256
ZnCl2 + 2NaOH = Zn(OH)2 + 2NaCl.
Zn(OH)2 (белого цвета) в воде практически не растворяется. Имеет имеет
амфотерный характерный характер. Растворяется в кислотах:
Zn(OH)2 + 2HCl = ZnCl2 + 2Н2О,
Zn(OH)2+2HF = ZnF2↓+2H2O,
и в растворах щелочей:
Zn(OH)2 + 2КОН = K2[Zn(OH)4].
Соли цинка. Соли кислородсодержащих кислот – нитрат цинка
Zn(NO3)2, сульфат цинка ZnSO4, сульфит цинка ZnSO3, перхлорат цинка ZnClO4, ацетат цинка Zn(СН3СОО)2 – растворимы в воде. Из галогенидов цинка фторид цинка ZnF2 представляет собой вещество с ионной связью, другие галогениды (хлорид, бромид, йодид цинка) имеют ковалентный характер. Все галогениды цинка растворяются в воде, кроме фторида цинка.
Водные растворы солей цинка гидролизованы и имеют кислый характер, так как гидроксид цинка – слабое основание:
2ZnSO4 + 2Н2O ↔ (ZnOH)2SO4 + H2SO4
или в ионной форме: 2Zn2+ + 2Н2O ↔2(ZnOH)+ + 2H+
ZnCl2+ H2O ZnOHCl + HCI,
ZnS + 2HCl ZnCl2 + H2S,
(ZnOH)2CO3 + 4HI 2ZnI2 + CO2 + 3H2O,
(ZnOH)2CO3 2ZnO + CO2 +H2O.
Катион цинка Zn2+ – хороший комплексообразователь, чаще всего проявляет КЧ = 4.
ZnO + 2HNO3 + 5H2O = [Zn (OH2)6] (NO3)2.
Наиболее важными являются амминокомплексы:
Zn(OH)2 + 4NH3 = [Zn(NH3)4] (OH)2,
ZnSO4 + 4NH3 = [Zn(NH3)4]SO4.
257
Роль меди и цинка в функционировании живых организмов. Токсичность кадмия и ртути.
Все соединения элементов группы IВ (особенно Сu) и IIВ ядовиты. Ряд соединений меди используется для борьбы с с-х вредителями. На
основе оксидов меди получены сверхпроводящие керамические материалы. Цинк и кадмий находят широкое применение в производстве ряда важных сплавов (латунь, томпак, нейзильбер, подшипниковые сплавы), для защиты металлов от коррозии. Ртуть используется как катод при электрохимическом получении щелочей и хлора, в амальгамной металлургии для получения ряда металлов (Tl, Ga, In, Pb и др.) и сплавов тугоплавких металлов, для изготовления ламп дневного света, ртутных манометров и термометров.
Медь важна для всех форм жизни, хотя встречается в организмах в небольших количествах. Она является важным микроэлементом, необходимым для нормальной жизнедеятельности организмов. Комплексные соединения меди (ΙΙ) с биолигандами участвуют в метаболических процессах, особенно в окислительно-восстановительных реакциях. Медь входит в состав белков и ферментов, которые регулируют такие жизненноважные процессы, как кроветворение, тканевое дыхание, рост и развитие организма, окислительно-восстановительные процессы. При недостатке меди в организме содержание церулоплазмина снижается, что может стать причиной анемии.
Цинк является уникальным микроэлементом, входит в состав активных центров ферментов, которые катализируют реакции гидролиза, окислительно-восстановительные реакции, реакции переноса групп, реакции конденсации и изомеризации. Помимо каталитической функции цинк выполняет также структурную функцию (стабилизирует третичную структуру белка) и регуляторную функцию. Цинк играет важную роль в работе генетического аппарата клетки, так как входит в состав ДНК- и РНКполимераз, стабилизирует структуру ДНК и РНК.
В медицинской практике применяются оксид ZnO и сульфат цинка ZnSO4∙7Н2O местно как антисептические, вяжущие и подсушивающие средства. Сульфат цинка назначается в виде растворов и глазных капель. Оксид цинка применяется в составе различных мазей, паст, линиментов, свечей. Для профилактики и лечения цинкдефицитных состояний используются для приема внутрь сульфат цинка, глюконат цинка, аспартат цинка. Сульфат цинка включается в состав минерало-витаминных комплексов как источник микроэлемента цинка.
Ртуть и его соединения чрезвычайно токсичны для всех организмов и не имеют биологического значения. Особенно большую опасность представляют даже в малых дозах соли Cd и Hg (в частности катиона Hg2+). Высокой токсичностью обладают и некоторые органические соединения ртути.
Биологическая роль ртути отсутствует, a кадмия – не изучена.
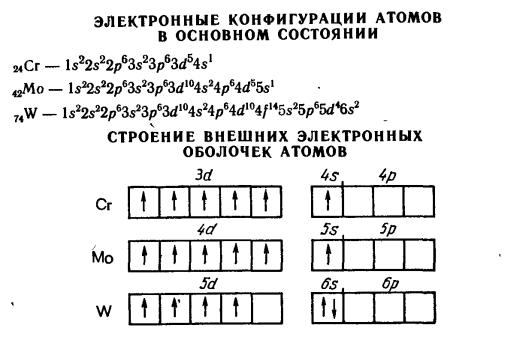
258
2.7.3 Общая характеристика элементов группы хрома
К d-элементам VIВ группы относятся металлы хром, молибден и вольфрам. Металлы подгруппы хрома встречаются преимущественно в виде руд: FeCr2O4 (хромит); PbCrO4(крокоит); MgCr2O4 (магнохромит); MoS2(молибденит); CaMoO4(повеллит); Fe2(MoO4)3×7,5H2O (молибдит); CaWO4(шеелит); (Fe, Mn) WO4 (вольфрамит).
Проявляемые степени окисления:
Элемент |
Cr |
Mo |
W |
Степени окисления |
+2,+3, +6 |
+2,+3,+4,+5,+6 |
+2,+3,+4,+5,+6 |
Для данных элементов, как и для всех d-элементов характерна закономерность: радиусы атомов сверху вниз в подгруппе увеличиваются, но незначительно. Поскольку масса атомов в том же ряду сильно возрастает, то это приводит к уплотнению электронных оболочек у молибдена и особенно у вольфрама. Вырвать электрон из тахой уплотненной структуры труднее, поэтому энергия ионизации при переходе от хрома к вольфраму возрастает, вследствие чего химическая активность элементов сверху вниз в подгруппе уменьшается. Ввиду того, что молибден и вольфрам имеют примерно одинаковый атомный и ионный радиусы, по свойствам они ближе друг к другу, чем к хрому.
Так же для элементов подгруппы Сг наблюдается общая для d – элементов закономерность: повышение в группе сверху вниз устойчивой С.О. Поэтому окислительная способность соединений, где элементы проявляют высшую С.О., равную +6, сверху вниз в подгруппе уменьшается, так как устойчивость соединений в этом ряду увеличивается. Например, в ряду кислот:
Н2СrO4, Н2МоO4, H2WO4 устойчивость увеличивается. A окислительная способность уменьшается.
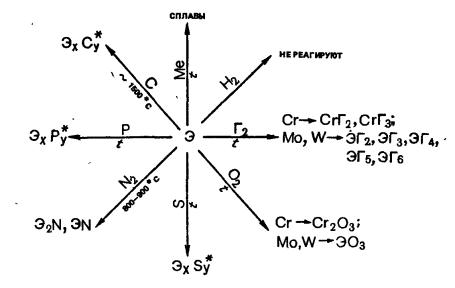
259
Для Сг, Mo, W наиболее типичны координационные числа 6 и 4. Известны также производные, в которых к.ч. Мо и W достигает 8.
Примеры: [Cr(OH)4]2- [Cr(H2O)6]3+,[MoF6]2-, [WF8]2-.
При этом в образовании связей могут участвовать d-орбитали предвнешнего уровня, а также s- и р-орбитали внешнего уровня.
Характер связи элементов подгруппы Сг в соединениях определяется во многом С.О. элемента. Для Cr, Mo, W при низких С.О. (+2) характерны ионные связи, а при высоких С.О. (+5, +6) – ковалентные связи.
Всоответствии с этим Сг+2O – основной оксид, Сг2+3О3 – амфотерный,
аСг+6О3 – кислотный.
Свойства простых веществ. Способы получения. Cr, Mo, W в
свободном виде – переработка природных соединений, конечными продуктами которой обычно являются оксиды Cr2O3, MoO3 и WO3. Окончательное выделение металлов осуществляется с помощью различных восстановителей при высоких температурах:
Cr2O3 + 2Al = 2Cr+ Al2O3,
MoO3 + 3С = Mo + 3СO,
WO3 + 3H2 = W + 3H2O.
Химические свойства. С химической точки зрения Cr, Mo и W малоактивны при обычных условиях и, несмотря на то, что в ряду напряжений находятся перед водородом, они практически не подвергаются коррозии из-за образования на их поверхности тонкой прочной пленки оксидов. С повышением температуры и при удалении защитной пленки они способны взаимодействовать со многими элементами. В ряду Cr–W химическая активность понижается, причем по большинству физических и химических свойств Mo и W схожи между собой и отличаются от Cr.
Реакции с простыми веществами
260
Реакции со сложными веществами
Cr + H2SO4 (разб) = CrSO4 + H2↑,
2Cr + 3H2O = Cr2O3 + 3H2↑,
W + 8HF + 2HNO3 = H2[WF8] + 2NO + 4H2O,
Mo + 3NaNO3 + 2NaOH (расплав) = Na2MoO4 + 3NaNO2 + H2O.
Соединения хрома и их особенности.
Оксиды. Элементы группы VIB образуют многочисленные оксидные соединения, соответствующие основным степеням окисления. Все оксиды при обычных условиях – твердые вещества. У хрома наиболее устойчивым является Cr2O3, а у Мо и W – MoO3 и WO3. В ряду Cr–W термодинамическая устойчивость кислотных оксидов ЭО3 растет.
Непосредственным взаимодействием металлов с кислородом могут быть получены лишь Cr2O3, MoO3 и WO3. Остальные оксиды получаются в результате различных химических процессов: разложения кислородсодержащих соединений, восстановления или окисления других оксидов и т.д.:
(NH4)2Cr2O7 = Cr2O3 + N2 + 4H2O,
K2Cr2O7 + H2SO4 = 2CrO3 + K2SO4 + H2O,
2MoO(OH)3 = Mo2O5 + 3H2O,
3MoO3 + 2NH3 = 3MoO2 + 3H2O + N2.
261
Низшие оксиды – сильные восстановители и проявляют основный характер. Черный оксид хрома (II) CrO получить очень трудно. CrO – неустойчивое соединение основного характера:
CrO + 2НС1 = СгСl2 + Н2О.
Рост степени окисления сопровождается усилением кислотных свойств. Так, Cr2O3– темно-зеленый порошок. Кристаллическая модификация Cr2O3
— черный порошок. Он отличается высокой тугоплавкостью, химически инертен. Cr2O3 – амфотерный оксид. В воде, кислотах и растворах щелочей не растворяется. Однако, при сплавлении его со щелочами и основными оксидами образуются соли метахромистой кислоты:
Cr2O3 + 2NaOH =2NaCrO2 + H2O,
Cr2O3 + 6KHSO4 = Cr2(SO4)3 +3K2SO4 +3H2O,
Cr2O3 + СаО = Са (CrO2)2.
При сплавлении Cr2O3 с дисульфатом калия образуется сульфат хрома (Ш):
1)3K2S2О7 = 3K2SО4 + 3SО3;
2)Cr2O3 + 3SО3 = Cr2(SO4)3.
Суммарно: Cr2O3 + 3K2S2О7 = Cr2(SO4)3+ 3K2SО4.
CrO3 (как и MoO3 и WO3) – типичный кислотный оксид со свойствами сильнейшего окислителя.
Единственный хорошо растворимый оксид – CrO3. Он при растворении в воде образует хромовую кислоту, а при увеличении концентрации раствора
– дихромовую:
CrO3 + H2O = H2CrO4;
H2CrO4 + CrO3 = H2Cr2O7.
MoO3 и WO3 плохо растворимы в воде и их кислотная природа проявляется лишь при растворении в щелочах:
2KOH + ЭO3 = K2ЭО4 + Н2О.
При нагревании СгО3 легко разлагается:
4СгО3 = 2Сг2О3 + 3О2.
262
Гидроксиды (таблица 2.7.3)
Таблица 2.7.3 – Гидроксиды и соли хрома
Степень окисления хрома |
Гидроксиды |
Соли |
|
+6 |
H2CrO4 |
Хроматы (VI), (CrO42-) |
|
H2Cr2O7 |
Дихроматы (Cr2O72-) |
||
|
|||
+5 |
|
Хроматы (V), (CrO43-) |
|
+4 |
nCrO2·mH2O |
Хроматы (IV), (CrO44-) |
|
+3 |
Cr2O3·nH2O или Cr(OH)3 |
Cr3+ хромиты, (CrO2-) |
|
+2 |
Cr(OH)2 |
Cr2+ |
|
Гидроксид Cr(OH)2 |
и соли Cr2+ – сильные восстановители, уже на |
||
воздухе окисляющиеся до соединений Cr3+. Является нерастворимым в воде желтым веществом, которое получают подщелачиванием растворов солей хрома (П):
CrСl2 + 2NaOH = Сг(ОН)2 + 2NaCl.
Гидроксид хрома (II) – слабое основание, имеет основной характер, т.е. взаимодействует только с кислотами и не растворяется в растворах щелочей:
Сг(ОН)2 + 2НСl = СгСl2 + Н2О.
Сг(ОН)2 образует ряд комплексов. Для хрома в С.О. +2 характерно координационное число 6. Например, галогениды хрома (II) поглощают газообразный аммиак, образуя аммиакаты:
CrСl2 + 6NH3 = [Cr(NH3)6]Cl2.
Гидроксид Cr(OH)3 осаждается из растворов солей Cr3+ в виде зеленовато-серого студенистого осадка:
CrCl3 + 3NaOH = Cr(OH)3↓ + 3NaCl.
Cr(OH)3 – амфотерен, при взаимодействии со щелочами образует гидроксохромиты типа Mn+[Cr(OH)n+3] (n=1, 2, 3 и растет с увеличением концентрации щелочи):
Cr(OH)3 + NaOH = Na[Cr(OH)4],
Cr(OH)3 + 3NaOH= Na3[Cr(OH)6].
При прокаливании эти соли обезвоживаются и переходят в безводные
хромиты:
Na[Cr(OH)4] = NaCrO2 + 2H2O.
При растворении Cr(OH)3 в кислотах образуются соответствующие соли Cr3+.
263
Соединения xpoмa (VI): хромовые кислоты и их соли. Наиболее известны гидроксидные производные Cr6+. Это прежде всего кислоты типа Н2ЭО4 и Н2Э2О7 и соответствующие им соли.
Хромовая НCrО4 и дихромовая Н2Cr2О7 кислоты средней силы существуют только в водных растворах, но соли, соответствующие им – желтые хроматы (анион Cr2O42-) и оранжевые дихроматы (анион Cr2O72-), устойчивы и могут быть выделены из растворов.
В кислой среде хромат-ион переходит в дихромат-ион:
Cr2O42- +2Н+ ↔ Н2О + Cr2O72-, и наоборот, под действием щелочи дихромат-ион превращается в хромат-ион: Cr2O72- + 2ОН- ↔ Н2О + 2 Cr2O42-
При нагревании дихроматы малоустойчивы:
K2Cr2O7 = 4 KCrO2 + 3О2.
Окислительные свойства соединений хрома (VІ). Дихроматы, как и хроматы, в кислой среде являются сильными окислителями за счёт восстановления Cr(VI) до Cr(III).
В нейтральной среде обычно образуется Cr(OH)3:
K2Cr2O7+3(NH4)2S + H2O = 2Cr(OH)3+ 3S + 6 NH3 + 2KOH,
В щелочной – гидроксохромиты:
2Na2CrO4+3(NH4)2S + NaOH + H2O = 2Na3[Cr(OH)6]+ 3S + 6 NH3.
Наибольшая окислительная активность хроматов проявляется в кислой среде (продукты восстановления – соли Cr+3):
K2Cr2O7 + 14HCl = 2CrCl3 + 3Cl2 + 2KCl +7Н2О.
Применение и роль в живой природе. Наиболее широкая область применения хрома, молибдена и вольфрама – металлургия, где эти металлы используются в качестве важнейших легирующих компонентов в производстве специальных сортов сталей и сплавов, придавая им высокую коррозионную стойкость, износоустойчивость, твердость и жаропрочность.
Хром широко используется для хромирования (получения гальванических защитных покрытий). Многие его соединения применяются как окислители (K2Cr2O7, CrO3) или восстановители (соли Cr2+) в различных химических производствах, в качестве абразивных материалов, катализаторов и т.д. Хром обладает важными биогенными свойствами. Являясь составной частью растительных и животных организмов, он участвует в деятельности ферментов (например, пепсина). При недостатке хрома замедляется рост животных, нарушается углеводный обмен, заболевают глаза. Растворимые соединения хрома ядовиты.
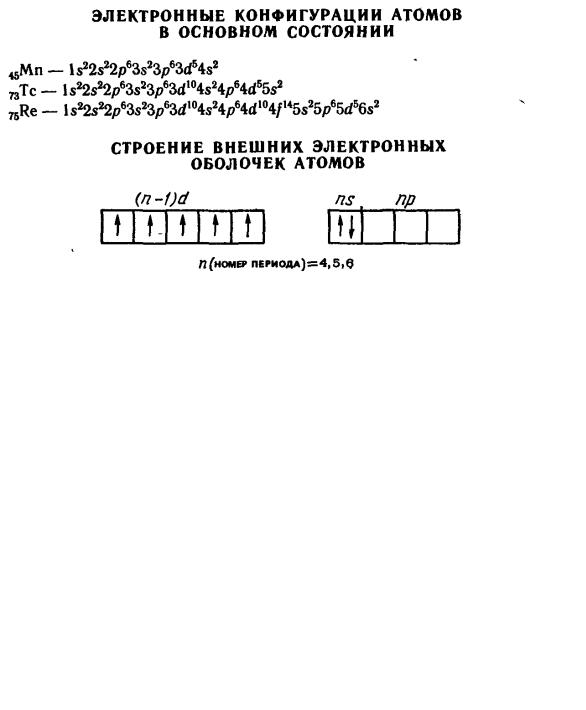
264
2.7.4 Общая характеристика элементов группы марганца
К d-элементам VIIB-группы относятся марганец, рений, техниций, борий. Наибольшее практическое значение имеют только марганец и его соединения. Mn выполняет важные функции в организме человека, являясь эссенциальным элементом.
Проявляемые степени окисления:
Элемент |
Mn |
Tc |
Re |
Степени окисления элемента в |
+2, +3+,4, |
+2, +4, |
+2, +3,+4, |
соединениях |
+6,+7 |
+6,+7 |
+6,+7 |
Свойства простых веществ представлены в таблице 2.7.4.1.
Таблице 2.7.4.1 – Физические свойства элементов группы марганца
Элемент |
Mn |
Tc |
Re |
|
|
|
|
Основная форма |
|
|
|
существования при обычных |
Металл |
Металл |
Металл |
условиях (в скобках тип |
(кубическая) |
(гексагональная) |
(гексагональная) |
кристаллической решетки |
|
|
|
Цвет в компактном |
|
|
|
состоянии |
Серебристый |
Серебристо-серый |
Серовато-белый |
В порошкообразном |
Светло-серый |
Серый |
Черный |
состоянии |
|
|
|
Плотность г/см3 (293 К) |
7,44 |
11,487 |
21,03 |
Температура плавления |
1244 |
2172 |
3180 |
|
|
|
|
Температура кипения |
1962 |
4877 |
5627 |
|
|
|
|
Химические свойства. Mn, Tc, Re взаимодействуют со многими реагентами, проявляя степень окисления от +2 до +7. Это взаимодействие особенно усиливается при нагревании или измельчении. Химические свойства элементов также в значительной степени зависят от их чистоты.
Химическая активность элементов понижается от Mn к Re (Mn в ряду
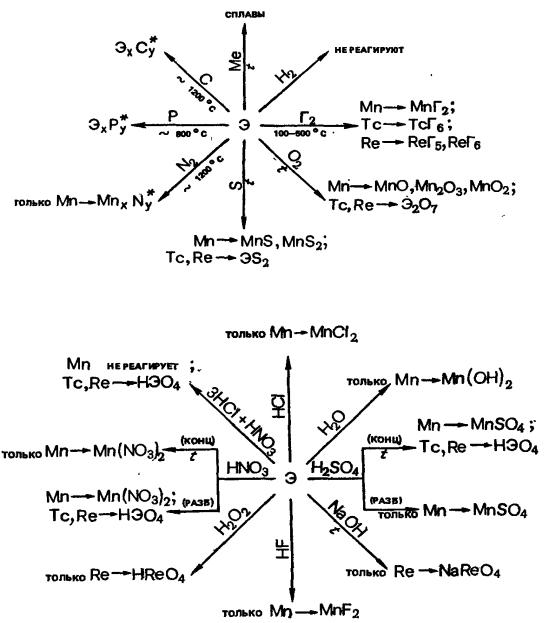
265
напряжений находится до водорода, а Те и Re-после него).
При комнатной температуре в порошкообразном состоянии все они окисляются во влажном воздухе. Металлический Mn в атмосфере сухого воздуха окисляется только с поверхности из-за образующейся тонкой и плотной пленки оксид.
Реакции с простыми веществами
Реакции со сложными веществами
Mn + 2H2O = Mn(OH)2 + H2,
Mn + H2SO4(разб) = MnSO4 + H2,
Mn + H2SO4(конц) = MnSO4 + SO2 + H2O, 3Tc + 7HNO3(разб) = 3HTcO4 + 7NO + 2H2O,
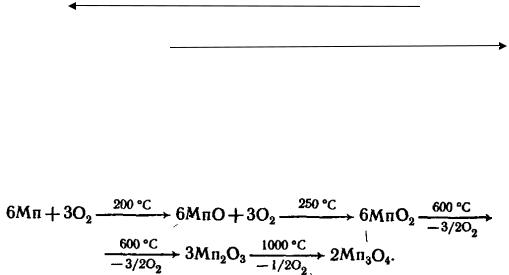
266
2Re + 7H2O2 = 2HReO4 + 6H2O.
Соединения марганца (ІІ), (IV), (VI) и (VII). Их формулы представлены в таблице 2.7.4.1.
Таблица 2.7.4.2 – Оксиды элементов группы марганца
Степень |
|
|
|
|
|
|
|
окисления |
+2 |
+3 |
+4 |
+5 |
+6 |
+7 |
|
элемента в |
|||||||
|
|
|
|
|
|
||
оксидах |
|
|
|
|
|
|
|
Mn |
MnO |
Mn2O3 |
MnO2 |
|
MnO3 |
Mn2O7 |
|
|
|
|
|
|
|
|
|
Tc |
|
|
TcO2 |
|
|
Tc2O7 |
|
|
|
|
|
|
|
|
|
Re |
ReO*H2O |
Re2O3*H2O |
ReO2 |
Re2O5 |
ReO3 |
Re2O7 |
|
|
|
|
|
|
|
|
|
|
|
Усиление основных свойств |
|
|
|||
Усиление кислотных свойств
Устойчивость оксидов ЭО, Э2О3, и ЭО2 в ряду Mn-Re уменьшается, а Э2О5, ЭО3 и Э2О7-наоборот, увеличивается.
Оксиды Мn в степенях окисления +2, +3,+4 могут быть получены непосредственным окислением элемента. Основная цепь термических превращений:
Обычные способы получения оксидов – термическое разложение солей и гидроксидов, восстановление или окисление соединений соответствующих степеней окисления:
Mn(OH)2 = MnO + H2O,
Mn(NO3)2 = MnO2 + 2NO2,
3Mn2O3 + KClO3 = 6MnO2 + KCl.
Оксид марганца(ΙΙ) MnO представляет собой основной оксид, практически нерастворимый в воде, легко растворимый в кислотах с образованием солей марганца(ΙΙ):
MnO + 2HCl = MnCl2 + H2О
267
Соответствующий этому оксиду гидроксид может быть получен действием щелочей на растворы солей Mn(ΙΙ):
MnCl2 + 2NaOH = Mn(OH)2↓+ 2NaCl
Гидроксид марганца (ΙΙ) – белый рыхлый осадок, на воздухе быстро окисляется и становится бурым, т.к. образуется гидроксид марганца (ΙV):
2Mn(OH)2 + 2H2О + O2 = 2Mn(OH)4↓
Гидроксид марганца (ΙΙ) является основанием, легко взаимодействует с кислотами и образует соли марганца(ΙΙ):
Mn(OH)2 + H2SO4 = MnSO4 + 2H2O
Соли марганца (ΙΙ). Важнейшими солями марганца(ΙΙ) являются дигидрат хлорида марганца MnCl2∙2H2O, гексагидрат сульфата марганца MnSO4∙6H2O, гексагидрат нитрата марганца Mn(NO3)2∙6H2O. Большинство солей марганца (ΙΙ) хорошо растворимо в воде. Водные растворы солей имеют кислую реакцию, так как подвергаются гидролизу.
2MnSO4 + 2H2O ↔ (MnОН)2SO4 + H2SO4
или в ионной форме: 2Mn2+ + 2H2O ↔ 2MnOН+ + 2H+
Оксид марганца(ΙV) MnO2 – самый устойчивый из всех оксидов марганца, является амфотерным оксидом. Однако кислотные и основные свойства выражены очень слабо. При сплавлении со щелочами оксид марганца(ΙV) ведет себя как кислотный оксид и образует манганит натрия:
MnO2 + 4NaOH = Na4MnO4 + 2H2O
На холоду оксид марганца(ΙV) не реагирует с разбавленными кислотами и не образует солей.
Для оксида марганца(ΙV) характерна окислительно-восстановительная двойственность. Оксида марганца(ΙV) является энергичным окислителем и реагирует с хлороводородной кислотой, окисляя ионы хлора:
MnO2 + 4HCl = MnCl2 + Cl2 + 2H2О
Может окислять в кислой среде соли железа(ΙΙ):
MnO2 + 2FeSO4 + 2H2SO4 = MnSO4 + Fe2(SO4)3 + 2H2O
268
В щелочной среде проявляет восстановительные свойства:
3MnO2 + KClO3 + 6KOH = 3K2MnO4 + KCl + 3H2O
При действии сильных окислителей окисляется сам:
3MnO2 + KClO3 + 6KOH 3K2MnO4 + KCl + 3H2O
3K2MnO4 + KCl + 3H2O
Гидроксид марганца(ΙV) Mn(OH)4 как и оксид марганца(ΙV) обладает амфотерными свойствами, при взаимодействии с кислотами образует нестойкие соли:
Mn(OH)4 + 4HCl = MnCl4 +4H2О
Mn(OH)4 + 2H2SO4 = Mn(SO4)2+ 4H2O
В водных растворах соли марганца (ΙV) нацело подвергаются гидролизу. При взаимодействии со щелочами образуются манганиты:
Mn(OH)4 + 4NaOH = Na4MnO4 + 4H2O
Оксид марганца(VΙΙ) Mn2O7, в отличие от других оксидов марганца, не является твердым веществом при обычных условиях, а представляет собой маслянистую жидкость темно-зеленого цвета.
Оксид марганца(VΙΙ) реагирует с водой на холоду, образуя марганцевую кислоту:
Mn2O7 + H2O = 2НMnO4
Марганцевая кислота относится к сильным кислотам. Соли этой кислоты называются перманганатами.
Наиболее известной является калиевая соль марганцевой кислоты – перманганат калия КMnO4. Он представляет собой темно-фиолетовые кристаллы, умеренно растворимые в воде. Растворы перманганата калия имеют темно-малиновый, а при больших концентрациях – фиолетовый цвет.
Растворы перманганата калия неустойчивы и медленно разлагаются. Процесс разложения ускоряется в присутствии кислоты:
4KMnO4 + 4Н3О+ = 3О2 + 4К+ + 6H2О + 4MnO2↓
В нейтральных и слабощелочных растворах в отсутствие света разложение протекает медленно. Это учитывают в аналитической практике при приготовлении стандартных растворов перманганата калия.
При нагревании перманганаты разлагаются с выделением кислорода:
2KMnO4 = K2MnO4 + MnO2 + О2
269
Перманганат калия – сильный окислитель, восстанавливается в зависимости от рН среды в различные продукты. В кислой среде образует ион марганца Mn+2, в нейтральной среде – диоксид марганца MnO2, в щелочной среде – манганат-ион MnO42-. Например, перманганат калия в сернокислой среде переводит соли Fe(ΙΙ) в соли Fe(ΙΙΙ):
2KMnO4 + 10FeSO4 + 8H2SO4 = 2MnSO4 + 5Fe2(SO4)3 + K2SO4 + 8H2O
Являясь сильным окислителем, перманганат калия выделяет из соляной кислоты хлор:
2KMnO4 + 16HCl = 2MnCl2 + 5Cl2 + 2КCl + 8H2О
Перманганат калия реагирует в кислой среде с раствором пероксида водорода с выделением молекулярного кислорода:
2KMnO4 + 5Н2О2 + 3H2SO4 = 2MnSO4 + 5O2 + K2SO4 + 8H2O
В нейтральной среде в присутствии сульфита натрия фиолетовый раствор перманганата калия обесцвечивается, выпадает бурый осадок диоксида марганца, и раствор приобретает щелочную реакцию:
2KMnO4 + 3Na2SO3 + H2O = 2MnO2↓ + 3Na2SO4 + 2KOH
В щелочной среде перманганат калия восстанавливается в манганат зеленого цвета:
2KMnO4 + К2SO3 + 2KOH = 2К2MnO4 + К2SO4 + H2O
Значение марганца для жизнедеятельности живых организмов
Марганец – важнейший микроэлемент, входит в состав многих ферментов, катализирующих окислительно-восстановительные реакции, реакции с участием АТФ и др. Марганец усиливает иммунный ответ организма на действие чужеродных белков, ускоряя синтез антител.
Марганец оказывает влияние на многие метаболические процессы, однако не все механизмы этого влияния достаточно хорошо изучены.
Соли марганца включают в состав различных витаминно-минеральных комплексов как источники микроэлемента марганца.
В медицинской практике применяется перманганат калия в виде растворов как антисептическое средство.
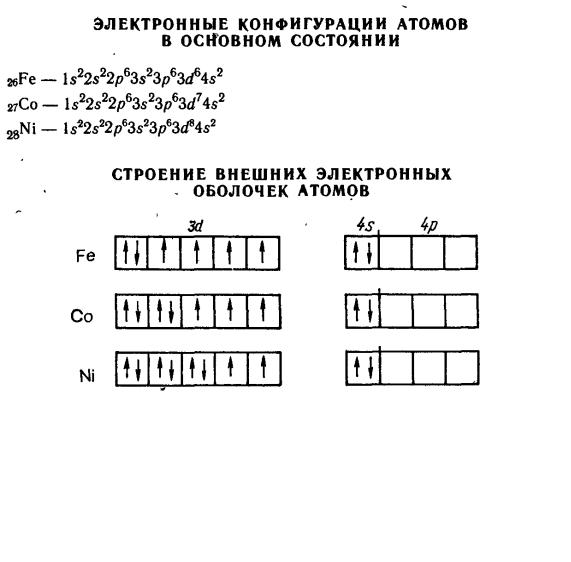
270
2.7.5 Общая характеристика элементов семейства железа
В VIIIB-группу входят девять d-элементов, которые подразделяют на три подгруппы, сходные по свойствам:
•подгруппу железа (железо Fe, рутений Ru, осмий Os);
•подгруппу кобальта (кобальт Co, родий Rh, иридий Ir);
•подгруппу никеля (никель Ni, палладий Pd, платина Pt).
Особую близость свойств проявляют пары элементов рутений-осмий, родий-иридий и палладий-платина, поэтому их объединяют в семейство
платиновых металлов.
Железо – один из самых распространенных элементов земной коры. Основные минералы железа представлены оксидами, сульфидом и карбонатом.
Оксидные минералы часто называют железняками: магнитный железняк (магнетит) Fe3O4, красный железняк (гематит) Fe2O3, бурый железняк (лимонит) FeO(OH). Сульфидный минерал FeS2 называется пиритом, карбонатный минерал FeCO3 – сидеритом.
Проявляемые степени окисления:
Элемент |
Fe |
Co |
Ni |
|
Степени окисления элемента |
+2,+3,+6 |
+2,+3 |
+2,+3,+4 |
|
в соединении |
||||
|
|
|
||
Свойства простых веществ представлены в таблице 2.7.5. |
||||
Таблице 2.7.5 – Физические свойства элементов группы марганца |
||||
Элемент |
Fe |
Co |
Ni |
|
Основная форма |
Металл |
Металл |
Металл |
|
существования при обычных |
(кубическая |
(гексагональная) |
(кубическая |
|
271
условиях (тип крист. решетки) |
объемноцентр |
|
гранецентрированн |
|
|
ированная) |
|
ая) |
|
Цвет (порошкообразное |
Серый |
Серый |
Серый |
|
состояние) |
||||
|
|
|
||
Плотность p, г/см³ (293 К) |
7,874 |
8,84 |
8,90 |
|
, С |
1535 |
1493 |
1453 |
|
, С |
2750 |
2870 |
2732 |
Химические свойства. Железо, кольбат и никель относятся к металлам средней химической активности, причем в ряду Fe–Ni она понижается. Fe, Co, Ni взаимодействуют при нагревании со многими неметаллами. Особенно легко это взаимодействие протекает во влажном воздухе и если Fe, Co, Ni находятся в мелкораздробленном состоянии.
С разбавленными кислотами Fe, Co, (Ni при нагревании) реагируют с выделением водорода. Концентрированные H2SO4 и HNO3 пассивируют поверхность металлов, но при нагревании взаимодействие происходит. Со щелочами (даже в расплавах) металлы не реагируют.
Реакции с простыми веществами
Реакции со сложными веществами
В раскаленном состоянии порошок железа легко сгорает с образованием оксида железа(ΙΙΙ):
4Fe + 3O2 = 2Fe2O3
272
В присутствии следов влаги легко окисляется хлором, образуя хлорид железа(ΙΙΙ):
2Fe +3Cl2 = 2FeCl3
При высокой температуре раскаленное железо реагирует с водой, что используется для получения водорода (железо-паровой метод):
3Fe + 4Н2О = Fe3O4 + 4Н2,
Ni + H O ↔NiO + H .
Железо относится к металлам со средней восстановительной активностью. При комнатной температуре легко взаимодействует с разбавленными кислотами, вытесняя водород:
Fe + 2НСl = FeСl2 + Н2,
Fe + Н2SO4 = FeSO4 + Н2
Концентрированные серная и азотная кислоты пассивируют железо. При нагревании пассивирование снимается, и железо окисляется до солей железа (ΙΙΙ):
2Fe + 6H SO (конц.) = Fe (SO ) + 3SO + 2H O, 4Fe + 10HNO (разб.) = 4Fe(NO ) + NH NO + 3H O,
3Ni + 8HNO (разб.) = 3Ni(NO ) + 2NO + 4H O.
Железо восстанавливает металлы, стоящие в ряду стандартных электродных потенциалов правее его. Например, железо осаждает медь из раствора сульфата меди:
Fe + CuSO4 = FeSO4 + Cu.
Оксиды, гидроксиды и соли железа, кобальта и никеля. Комплексные соединения железа и кобальта.
Оксиды. Fe, Co, Ni образуют как простые оксиды типа ЭО и Э О , так и смешанные – Э О , причем Ni O и Co O малоустойчивы. Все оксиды – твердые вещества, с большей долей нестихиометрии, практически нерастворимы в воде и щелочах, что свидетельствует об основном характере проявляемых свойств (только Fe O , и в некоторой степени Co O – амфотерны).
273
Оксиды ЭО. Эти оксиды (FeO – черного цвета, СоО – серо-зеленого цвета и NiO — зеленого цвета) практически не растворимы в воде. Оксиды образуются как при непосредственном окислении металлов, так и в результате различных процессов разложения карбонатов, нитратов,
гидроксидов, например: 2Fe(NO ) = 2FeO + 4NO + O .
Оксиды типа ЭО могут быть получены также восстановлением оксидов Э О водородом или СО. При нагревании совместно с восстановителем (H , CO, C и др.) оксиды ЭО восстанавливаются до металлов, а при обычном нагревании переходят в оксиды типа Э О или Э О :
Fe (200°) → Fe O ,
CoO (100°) → Co O ,
Fe2О3 + Н2 = 2FeO + Н2О,
Fe2O3 + СО → 2FeO + СО2.
СоО и NiO получают разложением гидроксидов н карбонатов:
CoCО3 = СоО + СО2,
Со(ОН)2 = СоО + Н2О,
Ni(OH)2 = NiO + Н2О.
Оксид Со(II) образуется также при взаимодействии простых веществ:
2Со + O2 = 2СоО.
Оксиды FeO, СоО, NiO взаимодействуют легко с кислотами:
ЭО + 2НСl = ЭСl2 + Н2O.
FеО и СоО при кипячении растворяются еще в концентрированных растворах щелочей, проявляя слабо амфотерный характер:
СоО + 2NaOH + Н2O = Na2[Co(OH)4].
Оксиды Э2О3. Из этих оксидов при обычных условиях устойчив лишь Fe O (буро-красный порошок). Его получают при обжиге колчедана:
4FeS2 + 11O2 = 2Fe2O3 + 8SO2
274
Он также может быть получен обезвоживанием его гидрата:
2Fe(OH)3 = 3H2О + Fe2O3.
Кроме того, Fe O встречается в природе в виде гематита Fe O .
Со2О3 и Ni2О3 можно получить дегидратацией оснований в специальных условиях (t°): 2Со(ОН)3 = Со2О3 + 3Н2О.
Со2О3 и Ni2О3 – неустойчивые соединения, легко разлагаются. Например, 6Со2О3 = О2 + 4Со3О4.
Оксид Fe O проявляет амфотерные свойства с преобладанием основных свойств. Он взаимодействует со щелочами, оксидами и карбонатами различных металлов (обычно при сплавлении) с образованием ферритов – солей железистой кислоты HFeO , не выделенной в свободном состоянии:
Fe O + 2NaOH = 2NaFeO + H O, Fe O + Na2CО3 = 2NaFeО2 + СО2
Fe O растворяется в кислотах с образованием светло-фиолетовых аквакомплексов |Fe(ОН2)6]3+:
Fe O + 6HCl + 9Н2О = 2[Fe(OH2)6]Cl3.
Со2О3 и Ni2О3, растворяясь в кислотах, проявляют окислительные свойства, окисляя соляную кислоту до хлора, а из кислородсодержащих кислот выделяют кислород.
Ni2О3+ 6НCl = 2NiCl2 + Cl2 + 3H2О.
Гидроксиды и их производные Гидроксиды Э(ОН)2. При добавлении щелочей к растворам,
содержащим Э² , выпадают в осадок гидроксиды Э(ОН) . Гидроксид железа (II) желтовато-белого цвета, на воздухе легко превращается в красноватокоричневый Fe(OH) :
4Fe(OH) + O + 2H O = 4Fe(OH) .
4Со(ОН)2 + О2 + 2Н2О = 4Со(ОН)3.
Гидроксиды Э(ОН) легко растворимы в кислотах, но под действием сильно концентрированных щелочей образуют растворимые комплексные соединения.
Э(ОН)2 + 2НCl = ЭCl2+ 2Н2О,
275
2NaOH(конц.) + Со(ОН)2 = Na2[Co(OH)4],
4NaOH(конц.) + Fe(OH)2 = Na4[Fe(OH)6].
Ni(OН)2 в растворах щелочей не растворяется, проявляя лишь основные свойства.
Гидроктды Э(ОН)3. При действии щелочей на растворы солей железа(ΙΙΙ) образуется красно-бурый осадок гидроксида железа(ΙΙΙ):
FeСl3 + 3NaOH = Fe(OH)3↓+ 3NaCl
Fe(OH)3 обладает амфотерными свойствами с преобладанием основных свойств. При взаимодействии с разбавленными кислотами легко образует соответствующие соли:
Fe(OН)3 + 3НСl = FeСl3 + 3H2O,
2Fe(OН)3 + 3H2SO4 = Fe2(SO4)3 + 6H2O.
При длительном нагревании гидроксид железа(ΙΙΙ) за счет амфотерных свойств реагирует с концентрированными растворами щелочей с образованием гидроксокомплексов с К.Ч. 4 или 6:
Fe(OН)3 + NaOH = Na[Fe(OH)4],
Fe(OН)3 + 3NaOH = Na3[Fe(OH)6].
При сплавлении гидроксида железа(ΙΙΙ) с щелочами образуются ферриты. Образование ферритов указывает на амфотерность гидроксида железа(ΙΙΙ):
Fe(OН)3 + NaOH = NaFeO2 + 2H2O
Соли железа(ΙΙ). Важнейшими солями железа(ΙΙ) являются гептагидрат сульфата железа FeSO4∙7H2O (железный купорос), гексагидрат нитрата железа Fe(NO3)2∙6H2O. Кристаллогидраты имеют зеленое окрашивание. Большинство солей железа(ΙΙ) хорошо растворимо в воде. Водные растворы солей имеют кислую реакцию, так как подвергаются гидролизу:
2FeSO4 + 2H2O ↔ (FeОН)2SO4 + H2SO4
или в ионной форме: 2Fe2+ + 2H2O ↔ 2FeOН+ + 2H+
Катион железа(ΙΙ) обладает восстановительными свойствами и может окисляться различными окислителями. Например, сульфат железа(ΙΙ)
276
восстанавливает перманганат калия в сернокислой среде до сульфата марганца (ΙΙ):
10FeSO4 + 2КMnO4 + 8H2SO4 = 5Fe2(SO4)3 + 2MnSO4 + K2SO4 + 8H2O
Катион железа(ΙΙ) образует как катионные, так и анионные комплексы. Например, из катионных комплексов известны аквакомплексы [Fe(H2O)6]2+,
амминокомплексы [Fe(NH3)6]2+. Анионные комплексы – в большинстве своем нестойкие соединения. Наиболее устойчивым является цианидный комплекс, образующийся по реакции:
6КСN + FeSO4 = К4[Fe(СN)6] + K2SO4
Гексацианоферрат(ΙΙ) калия называется желтой кровяной солью, она широко применяется в аналитической химии для обнаружения иона Fe3+.
Для обнаружения иона Fe2+ применяется раствор гексацианоферрата(ΙΙΙ) калия (красная кровяная соль). В результате реакции образуется интенсивно синий осадок турнбулевой сини:
FeСl2 + К3[Fe(СN)6] = КFe[Fe(СN)6]↓ + 2КСl
Соли железа (ΙΙΙ). Хлорид железа(ΙΙΙ) представляет собой темнокоричневые кристаллы, расплывающиеся на воздухе вследствие гигроскопичности. Поглощая влагу из воздуха, хлорид железа превращается в кристаллогидраты и приобретает буро-оранжевую окраску.
Сульфат железа(ΙΙΙ) – очень гигроскопичные, расплывающиеся на воздухе белые кристаллы. Его кристаллогидрат Fe2(SO4)3∙9H2O имеет желтое окрашивание. Сульфат железа при взаимодействии с сульфатами щелочных металлов и аммония образует двойные соли – квасцы, например железоаммонийные: Fe2(SO4)3 + (NH4)2SO4 = 2Fe(NH4)(SO4)2
В разбавленном растворе соли железа(ΙΙΙ) сильно гидролизуются до основных солей в отличие от солей железа(ΙΙ). Это объясняется тем, что гидроксид железа(ΙΙΙ) – более слабое основание, чем гидроксид железа(ΙΙ).
FeСl3 + H2O ↔ FeОНСl2 + НСl,
FeОНСl2 + H2O ↔ Fe(ОН)2Сl + НСl,
Fe2(SO4)3 + 2H2O ↔ 2FeОН(SO4) + Н2SO4,
2FeОН(SO4) + 2H2O ↔ [Fe(ОН)2]2(SO4) + Н2SO4.
Благодаря гидролизу образующиеся гидроксо-ионы железа FeОН2+ и [Fe(ОН)2]+ окрашивают растворы солей железа(ΙΙΙ) в желто-бурый цвет. Ионы Fe3+ почти бесцветные. При подкислении окраска растворов солей
277
железа(ΙΙΙ) становится более светлой вследствие подавления гидролиза; при нагревании окраска темнеет, происходит усиление гидролиза.
Соединения железа(ΙΙΙ), в том числе и соли, проявляют окислительные свойства и восстанавливаются в соединения железа(ΙΙ). Например, окисляют раствор йодида калия до молекулярного йода:
2FeСl3 + 2КI = 2FeСl2 + I2 + 2КСl
Из комплексных соединений железа(ΙΙΙ) наибольшее значение имеет красная кровяная соль – гексацианоферрат(ΙΙΙ) калия К3[Fe(СN)6]. Эта соль широко применяется для обнаружения катиона Fe2+.
Для обнаружения иона Fe3+ используют гексацианоферрат(ΙΙ) калия (желтая кровяная соль). Образуюшийся осадок синего цвета называется берлинской лазурью:
FeСl3 + К4[Fe(СN)6] → КFe[Fe(СN)6]↓ + 3КСl
Для отличия катионов Fe2+ и Fe3+ рекомендуется реакция с раствором тиоцианата аммония. Соли железа(ΙΙΙ) образуют тиоцианат железа (ΙΙΙ) кроваво-красного цвета:
FeСl3 + 3NH4SCN → Fe(SCN)3 + 3NH4Сl
При взаимодействии с тиоцианатом аммония солей железа(ΙΙ) раствор остается бесцветным
Соединения железа(VΙ). Такие соединения железа крайне редки и немногочисленны. При нагревании оксида железа(ΙΙΙ) с нитратом и гидроксидом калия образуется феррат калия – соль железной кислоты:
Fe2O3 +4КОН + 3КNO3 = 2К2FeO4 + 3КNO2 + 2H2O
Образующийся феррат калия – кристаллическое вещество красного цвета, проявляет сильные окислительные свойства, превосходящие даже калия перманганат. Соответствующая ферратам железная кислота Н2FeO4 и ее ангидрид FeO3 в свободном состоянии не существуют.
Биологическая роль железа, кобальта и никеля. В живых организмах железо является важным микроэлементом, катализирующим процессы обмена кислородом (дыхания). В организме взрослого человека содержится около 3,5 г железа (около 0,02%), из которых 75% являются главным действующим элементом гемоглобина крови, остальное входит в состав ферментов других клеток, катализируя процессы дыхания в клетках. Недостаток железа проявляется как болезнь организма (хлороз у растений и анемия у животных).
278
Обычно железо входит в ферменты в виде комплекса, называемого гемом. В частности, этот комплекс присутствует в гемоглобине и именно он окрашивает кровь в характерный красный цвет.
Неорганические соединения железа встречается в некоторых бактериях, иногда используется ими для связывания азота воздуха.
Кобальт, один из микроэлементов, жизненно важных организму. Он входит в состав витамина В12 (кобаламин). Кобальт задействован при кроветворении, функциях нервной системы и печени, ферментативных реакциях. Потребность человека в кобальте 0,007–0,015 мг, ежедневно. В теле человека содержится 0,2 мг кобальта на каждый килограмм массы человека. При отсутствии кобальта развивается акобальтоз.
Никель относится к числу микроэлементов, необходимых для нормального развития живых организмов. Однако о его роли в живых организмах известно немного. Известно, что никель принимает участие в ферментативных реакциях у животных и растений.
279
РАЗДЕЛ 3 ОРГАНИЧЕСКАЯ ХИМИЯ
3.1 ПРЕДМЕТ ОРГАНИЧЕСКОЙ ХИМИИ
ПЛАН
3.1.1Характеристика органических веществ.
3.1.2Формирование и основные положения теории строения органических соединений.
3.1.1 Характеристика органических веществ
Состав органических веществ, элементы-органогены Органическая химия – это раздел химической науки, в котором
изучаются соединения углерода – их строение, свойства, способы получения и практического использования. Соединения, в состав которых входит углерод, называются органическими.
Кроме углерода, органические вещества почти всегда содержат водород, довольно часто – кислород, азот и галогены, реже – фосфор, серу и другие элементы.
Органические соединения – это углеводороды (соединения углерода с водородом) и их производные.
Органические соединения отличаются от неорганических рядом характерных особенностей:
•почти все органические вещества горят или легко разрушаются при нагревании с окислителями, выделяя СО2;
•в молекулах органических соединений углерод может быть соединен почти с любым элементом Периодической системы;
•органические молекулы могут содержать последовательность атомов углерода, соединенных в цепи (открытые или замкнутые);
•молекулы большинства органических соединений не диссоциируют на достаточно устойчивые ионы;
•реакции органических соединений протекают значительно медленнее и в большинстве случаев не доходят до конца;
• |
среди органических соединений |
широко распространено |
явление изомерии; |
|
|
• |
органические вещества имеют |
более низкие температуры |
фазовых переходов (Ткип., Тпл.). |
|
|
Многообразие органических веществ
Благодаря особым свойствам элемента углерода, органические соединения очень многочисленны. Сейчас известно свыше 20 миллионов синтетических и природных органических веществ, и их число постоянно возрастает.
Принципы существования многообразия органических веществ:
280
1.Способность атомов углерода образовывать прочные связи с другими атомами углерода, что приводит к формированию цепей и циклов;
2.Способность атомов углерода соединяться с четырьмя различными атомами, что позволяет углеродным цепям разветвляться и образовывать производные соединения (галогенпроизводные, нитропроизводные и т.д.).
3.Вследствие разного вида гибридизации атомы углерода способны образовывать простые (С – С) связи, двойные (С = С), и тройные связи.
Место органической химии в системе естественных наук
Органическая химия имеет исключительно важное познавательное и народнохозяйственное значение:
• Природные органические вещества и их превращения лежат в основе явлений жизни. Поэтому органическая химия является химическим фундаментом биологической химии и молекулярной биологии – наук, изучающих процессы, происходящие в клетках организмов на молекулярном уровне. Исследования в этой области позволяют глубже понять суть явлений живой природы.
• Большое количество синтетических органических соединений производится промышленностью для использования в самых разных отраслях человеческой деятельности: полимерные материалы (каучуки, пластмассы, волокна, пленки, лаки, клеи и т.д.), поверхностно-активные вещества, красители, средства защиты растений, лекарственные препараты, вкусовые и парфюмерные вещества и т.п. Без знания основ органической химии современный человек не способен экологически грамотно использовать все эти продукты цивилизации.
Источники органических соединений
Сырьевыми источниками органических соединений служат: нефть и природный газ, каменный и бурый угли, горючие сланцы, торф, продукты сельского и лесного хозяйства.
Природный газ является смесью разнообразных углеводородов, основные компонентом которой является метан (95%) и в пределах 2% иных углеводородов (этана, пропана, бутана и др.)
Нефть – маслянистая, темная жидкость, легче воды и в ней нерастворима. По химическому составу это смесь углеводородов различных классов – предельные углеводородов, циклоалканов и ароматических.
Нефть – основное сырье для химической и нефтехимической промышленности. Главный способ ее переработки – фракционная перегонка. Она осуществляется в так называемых ректификационных колоннах. При обычном давлении нефть разделяется на три фракции: бензин (30–180 ᴏС), керосин (180–300 ᴏС) и мазут (остаток от перегонки). Из этих основных фракций нефти выделяют более узкие фракции: петролейный (нефтяной)
эфир (30–80 ᴏС), лигроин (110–140 ᴏС), уайт-спирит (150–210 ᴏС).
281
Из мазута перегонкой под уменьшенным давлением или с водяным паром получают соляровое масло, смазочные масла, вазелин, твердый парафин, а также битум, используемый при строительстве дорог.
Фракции нефти подвергаются очистке от сернистых и химически нестойких соединений с помощью серной кислоты, растворов щелочей, гипохлоритов.
С целью повышения выхода бензина проводят вторичную переработку нефтепродуктов – их крекинг. При высокой температуре (450–550 °С) и давлении осуществляют термический крекинг; в присутствии катализаторов, например, алюмосиликатов, – каталитический крекинг. При крекинге происходит «дробление» крупных молекул на более мелкие. Важным способом переработки нефтепродуктов является риформинг – превращение алканов и циклоалканов в ароматические углеводороды. При риформинге получают высококачественные сорта бензина.
Различают два основных типа крекинга-термический и каталитический.
Термический крекинг проводят при температуре 470–650 °С и давлении до 7 МПа. Углеводороды с большой молекулярной массой при крекинге превращаются в более ценные продукты - предельные и непредельные углеводороды с более низкой молекулярной массой. Например, при крекинге бутана можно получить:
Строение продуктов крекинга определяется структурой исходного алкана и условиями проведения крекинга. Термический крекинг – свободно радикальный процесс.
Продукты крекинга разделяют на ректификационной колонне. Наиболее ценная жидкая фракция - бензиновая. Бензин, полученный при крекинге, имеет более высокое октановое число, чем бензин прямой перегонки нефти. Однако химическая стойкость такого бензина невысокая, так как в его состав входят алкены, которые со временем окисляются и образуют смолообразные продукты.
Каталитический крекинг протекает в присутствии катализаторов (А1С13, Сr2О3, алюмосиликаты) при температуре 470–500°С и давлении 0,01– 0,1 МПа. Каталитическому крекингу подвергают в основном дизельную
282
фракцию. При этом происходит не только разрыв углеводородных цепей в молекуле (как при термическом крекинге), но и процессы изомеризации – превращение неразветвленных углеводородов в разветвленные. Это способствует образованию высокооктанового горючего.
Каталитический крекинг – более прогрессивный метод переработки нефтяного сырья, чем термический.
В результате каталитического крекинга образуется смесь жидких и газообразных углеводородов, которые разделяют на ректификационных колоннах. Газы каталитического крекинга содержат предельные (пропан и бутан) и непредельные (пропен и бутен) углеводороды. После разделения их используют для синтеза разнообразных органических соединений.
Пиролиз и риформинг. Если продукты нефтепереработки нагревать до температуры 650-700 °С при обычном давлении, то такой процесс называют пиролизом. При этом также происходит разрыв длинных углеродных цепей на более короткие. В результате увеличивается выход газообразных продуктов.
Бензин, полученный перегонкой нефти, можно превратить в высокосортный путем каталитического риформинга. Он протекает при
500°С в присутствии платинового катализатора. Если при крекинге в качестве сырья используют различные фракции нефти (от лигроина до мазута), то при риформинге сырьем служат низкооктановые бензины или лигроины, из которых получают высокооктановые бензины и ароматические углеводороды (в основном бензол).
Пиролиз и каталитический риформинг являются разновидностью крекинга нефтепродуктов.
Каменный уголь и другие виды ископаемых углей. В настоящее время в промышленности используется несколько путей переработки каменного угля: сухая перегонка (коксование, полукоксование), гидрирование, неполное сгорание, получение карбида кальция.
Сухая перегонка угля применяется для получения кокса или бытового газа. При коксовании угля получаются кокс, каменноугольная смола, надсмольная вода и газы коксования. Газы коксования содержат аммиак, водород, оксид и диоксид углерода, метан, этилен и другие углеводороды, в том числе ароматические. Выход каменноугольной смолы невелик – всего около 3%. Однако она вырабатывается в огромных количествах, так как масштабы коксохимического производства, обслуживающего металлургию, очень велики.
Каменноугольная смола представляет собой сложную смесь многих, преимущественно ароматических, углеводородов, кислород- и азотсодержащих веществ. Каменноугольная смола разделяется перегонкой на пять фракций: 1) до 170 °С – легкое масло, состоит преимущественно из углеводородов; 2) 170–230 °С – среднее масло, содержит в больших количествах фенолы; 3) 230–270 °С – тяжелое масло, из него выделяют нафталин; 4) 270–340 °С – антраценовое масло; 5) остаток – пек.
283
Важнейшими отраслями промышленности, которые производят органические вещества или перерабатывают органическое сырье, являются: производство каучука, резины, пластмасс, волокон, нефтехимическая промышленность, пищевое, фармацевтическое, лакокрасочное производства.
В наш век исключительно большое значение приобрело промышленное производство высокомолекулярных соединений. Природные органические соединения, в частности, нуклеиновые кислоты белки, гормоны, витамины играют основополагающую роль в жизнедеятельности растительных и животных организмов.
3.1.2 Формирование и основные положения теории строения органических соединений
Теория химического строения в своей основе была создана в 60-х годах XIX века. Если в первой половине XIX века основная задача органической химии состояла в изучении состава и свойств природных соединений, а также в разработке способов рационального их использования, то далее, в связи с развитием промышленности и ростом городов, к органической химии стали предъявлять все больше требований: требовались красители, лекарства, новое освещение, новые методы переработки пищевых продуктов и т.д.
Развитие органической химии тормозилось отставанием теоретических представлений. Не было единой химической символики.
Основой современной органической химии является теория строения органических соединений, созданная на базе теории химического строения А.М. Бутлерова и электронных (квантовохимических) представлений о строении атома и природе химической связи.
Основные положения теории:
1. Атомы, образуя молекулу органического вещества, располагаются не беспорядочно, а в строгой и определенной последовательности. Между атомами углерода возникает ковалентная связь. Она может быть одинарной (σ-связь), двойной (1σ и 1π – связи), тройной (1σ и 2π – связи).
Ковалентная связь характеризуется: длиной, энергией, полярностью связи. Между атомами С и Н тоже возникает σ-связь. К цепи, состоящей из двух атомов углерода, может присоединиться третий, четвертый атом и т.д., т.е. образуются углеродные цепи, причем их направленность в пространстве определяется направленностью электронного облака. Цепи могут быть
линейные, разветвленные и циклические. Во всех этих цепях атом углерода
четырехвалентен.
2.Свойства вещества зависит не только от того, какие атомы и
вкаком количестве входят в состав его молекулы, но и от того, в каком порядке они соединены между собой, т.е. от химического строения молекулы. Согласно этому положению теории, было обнаружено много органических веществ, обладающих одинаковым составом, но разными
284
свойствами. Это явление, открытое в 1830 году, было названо изомерией, а вещества с одинаковым составом – изомерами.
3.Атомы или группы атомов, образуя молекулу, взаимно влияют друг на друга, от чего зависит реакционная способность молекулы. Такое влияние особенно велико, если атомы непосредственно связаны друг с другом. Влияние существует и в том случае, если связь осуществляется через какой-то другой элемент. В этом случае важно учитывать такое явление, как перераспределение в молекуле электронной плотности (индуктивный эффект, эффект сопряжения).
4.Зная свойства вещества, можно установить его строение, и, наоборот, химическое строение вещества определяет его свойства.
Электронные представления в органической химии. Применение электронной теории строения атома и химической связи в органической химии явилось одним из важнейших этапов развития теории строения органических соединений. Именно эти представления дают возможность понять способы передачи взаимного влияния атомов в молекулах (электронные и пространственные эффекты) и поведение молекул в химических реакциях.
Согласно современным представлениям свойства органических соединений определяются:
• природой и электронным строением атомов;
• типом атомных орбиталей и характером их взаимодействия;
• типом химических связей;
• химическим, электронным и пространственным строением молекул.
Сегодня сущность теории можно выразить так: физические и
химические свойства органических соединений определяются составом их молекул, а также химическим пространственным и электронным строением.
Современная теория строения позволяет предсказывать основные химические и физические свойства органических соединений, исходя из их химического, пространственного и электронного строения. Теория строения играет ключевую роль в изучении и систематизации огромного фактического материала органической химии
Структурные формулы как средство отображения строения органических соединений
Структурная формула — изображение химических связей между атомами в молекуле с учетом их валентности (обозначается черточкой).
Способы изображения органических соединений:
a)Эмпирическая или молекулярная брутто-формула – C2H6;
б) |
Электронная молекулярная формула – |
; |
285
в) Структурная молекулярная формула –
г) ПолуструктурнаЯ молекулярная формула: – СН3-(СН2)5-СН3; д) Рациональная (графическая, скелетная) молекулярная формула –
Основные типы структурных фрагментов органических молекул:
простые и кратные связи, углеродные цепи и циклы, функциональные группы.
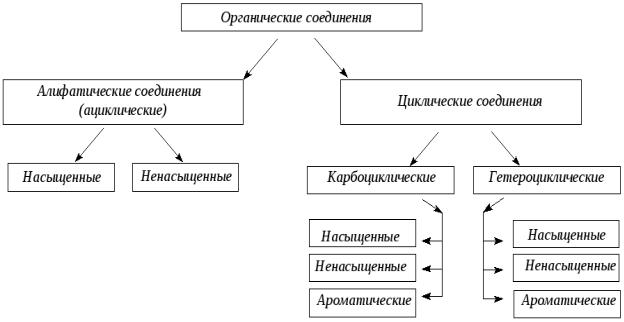
286
3.2 КЛАССИФИКАЦИЯ, НОМЕНКЛАТУРА, ИЗОМЕРИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПЛАН
3.2.1Классификация органических соединений.
3.2.2Номенклатуры органических соединений.
3.2.3Изомерия органических соединений.
3.2.1 Классификация органических соединений
Основные признаки классификации: скелет молекулы, степень насыщенности, наличие функциональных групп
Для классификации органических соединений по типам и построения их названий в молекуле органического соединения принято выделять углеродный скелет и функциональные группы. Углеродный скелет представляет собой последовательность химически связанных между собой атомов углерода. Функциональные группы образуют все атомы, кроме водорода, или группы атомов, связанные с атомами углерода.
По строению углеродного скелета органические соединения разделяют на ациклические (не содержащие циклов), циклические.
Ациклические соединения – соединения с открытой (незамкнутой) углеродной цепью. Эти соединения называются также алифатическими.
Ациклические соединения подразделяют также на соединения с
неразветвленной и разветвленной цепью. В этом случае учитывается число связей атома углерода с другими углеродными атомами. Атомы углерода могут образовывать между собой не только одинарные, но и кратные (двойные и тройные) связи. Соединения, содержащие только одинарные связи углерод – углерод, называют насыщенными; соединения с кратными углерод-углеродными связями называют ненасыщенными.
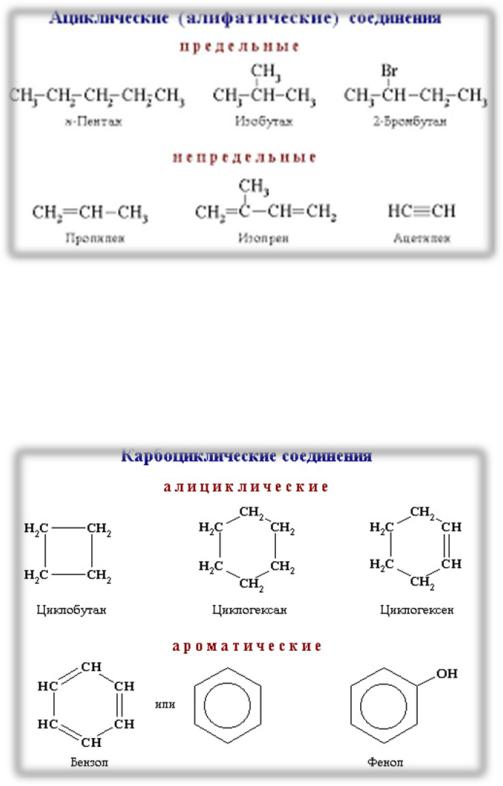
287
Циклические соединения – соединения с замкнутой углеродной цепью. В зависимости от природы атомов, составляющих цикл, различают
карбоциклические и гетероциклические соединения. Карбоциклические
соединения содержат в цикле только атомы углерода. Они делятся на две существенно различающихся по химическим свойствам группы:
алифатические циклические – сокращенно алициклические и ароматические
соединения.
В гетероциклическом скелете в углеродный цикл включается один или несколько гетероатомов, т.е. атомов, отличных от углерода. Чаще всего это кислород, сера и азот. Гетероатомы не рассматривают как функциональные группы, а считают их частью углеродного скелета.
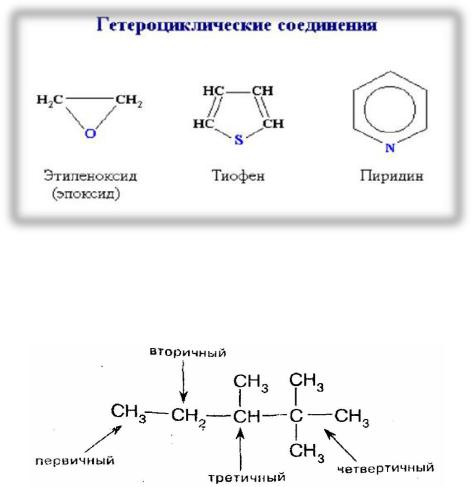
288
В самих углеродных скелетах классифицировать отдельные атомы углерода можно по числу химически связанных с ними атомов углерода. Если атом углерода связан с одним атомом углерода, то его называют
первичным, с двумя – вторичным, с тремя – третичным и с четырьмя – четвертичным:
Соединения, в которых кроме атомов углерода содержатся только атомы водорода, называют углеводородами. Однако в большинстве органических веществ, кроме атомов углерода и водорода, содержатся атомы других элементов. Эти атомы или их группировки, во многом определяющие химические и физические свойства органических соединений, называют
функциональными группами (таблица 3.2.1).
Соединения с одной функциональной группой называются монофункциональными (метанол СН3-ОН), с несколькими одинаковыми функциональными группами – полифункциональными (глицерин СН2(ОН)-СН(ОН)-СН2(ОН)), с несколькими разными функциональными группами – гетерофункциональными (молочная кислота СН3СН(ОН)-СООН).
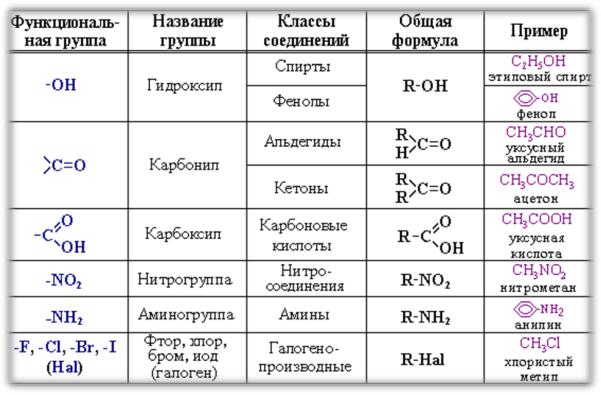
289
Таблица 3.2.1 – Наиболее характерные функциональные группы и соответствующие им классы соединений
3.2.2 Номенклатуры органических соединений
В настоящее время в органической химии общепринятой является систематическая номенклатура, разработанная Международным союзом чистой и прикладной химии (IUPAC). Наряду с ней сохранились и используются тривиальная и рациональная номенклатуры.
Тривиальные и систематические названия органических соединений
Тривиальная номенклатура состоит из исторически сложившихся названий, которые не отражают состава и строения вещества. Они являются случайными и отражают природный источник вещества (молочная кислота, мочевина, кофеин), характерные свойства (глицерин, гремучая кислота), способ получения (пировиноградная кислота, серный эфир), имя первооткрывателя (кетон Михлера, углеводород Чичибабина), область применения (аскорбиновая кислота). Преимуществом тривиальных названий является их лаконичность, поэтому употребление некоторых из них разрешено правилами IUPAC.
Систематическая номенклатура является научной и отражает состав, химическое и пространственное строение соединения. Название соединения выражается при помощи сложного слова, составные части которого отражают определенные элементы строения молекулы вещества.
Заместительная номенклатура IUРАС, основные принципы построения названий органических соединений
В основе правил номенклатуры IUPAC лежат принципы
290
заместительной номенклатуры, согласно которой молекулы соединений рассматриваются как производные углеводородов, в которых атомы водорода замещены на другие атомы или группы атомов. При построении названия в молекуле соединения выделяют следующие структурные элементы.
Родоначальная структура – главная цепь углеродная цепь или циклическая структура в карбо- и гетероциклах.
Углеводородный радикал – остаток формульного обозначения углеводорода со свободными валентностями (таблица 3.2.2.1).
Характеристическая группа – функциональная группа, связанная с родоначальной структурой или входящая в ее состав (таблица 3.2.2.2).
При составлении названия последовательно выполняют следующие правила.
1.Определяют старшую характеристическую группу и указывают
ееобозначение в суффиксе (таблица 3.2.2.2).
2.Определяют родоначальную структуру по следующим критериям в порядке падения старшинства:
а) содержит старшую характеристическую группу; б) содержит максимальное число характеристических групп; в) содержит максимальное число кратных связей; г) имеет максимальную длину.
Родоначальную структуру обозначают в корне названия в соответствии с длиной цепи или размером цикла: С1 – «мет», С2 – «эт», С3 – «проп», С4 – «бут» и далее – корни греческих числительных.
3.Определяют степень насыщенности и отражают ее в суффиксе: «ан» – нет кратных связей, «ен» – двойная связь, «ин» – тройная связь.
4.Устанавливают остальные заместители (углеводородные радикалы и младшие характеристические группы) и перечисляют их названия в префиксе в алфавитном порядке.
5.Устанавливают умножающие префиксы – «ди», «три», «тетра», указывающие число одинаковых структурных элементов (при перечислении заместителей в алфавитном порядке не учитываются).
6.Проводят нумерацию родоначальной структуры так, чтобы старшая характеристическая группа имела наименьший порядковый номер. Локанты (цифры) ставят перед названием родоначальной структуры, перед префиксами и перед суффиксами.
291
Таблица 3.2.2.1 – Названия алканов и алкильных радикалов, принятые систематической номенклатурой IUPAC
Алкан |
Название |
Алкильный |
Название |
|
радикал |
||||
|
|
|
||
CH4 |
Метан |
СН3- |
Метил |
|
|
|
|
|
|
CH3CH3 |
Этан |
CH3CH2- |
Этил |
|
|
|
|
|
|
|
|
CH3CH2CH2- |
Пропил |
|
CH3CH2CH3 |
Пропан |
|
|
|
|
Изопропил |
|||
|
|
|
||
|
|
|
|
|
|
|
CH3CH2СН2CH2- |
н-Бутил |
|
CH3CH2СН2CH3 |
н-Бутан |
|
|
|
|
втор-Бутил |
|||
|
|
|
||
|
|
|
|
|
|
|
|
Изобутил |
|
|
Изобутан |
|
|
|
|
|
|
||
|
|
|
трет-Бутил |
|
|
|
|
|
|
CH3CH2СН2CH2СН3 |
н-Пентан |
CH3CH2СН2CH2СН2- |
н-Пентил |
|
|
|
|
|
|
|
Изопентан |
|
Изопентил |
|
|
|
|
|
|
|
Неопентан |
|
Неопентил |
|
|
|
|
|

292
Таблица 3.2.2.2 – Названия характеристических групп (перечислены в порядке убывания старшинства)
Группа |
|
Название |
|
|
|
||
в префиксе |
в суффиксе |
||
|
|||
|
|
|
|
-(C)OOH* |
- |
овая кислота |
|
-COOH |
карбокси |
карбоновая кислота |
|
|
|
|
|
-SO3H |
сульфо |
сульфоновая кислота |
|
|
|
|
|
-(C)HO |
оксо |
аль |
|
|
|
|
|
-CHO |
формил |
карбальдегид |
|
|
|
|
|
>(C)=O |
оксо- |
он |
|
|
|
|
|
-ОН |
гидрокси |
ол |
|
|
|
|
|
-SH |
меркапто |
тиол |
|
|
|
|
|
-NH2 |
амино |
амин |
|
|
|
|
|
-OR** |
алкокси, арокси |
- |
|
-F, -Cl, -Br, -I |
фтор, хлор, бром, иод |
- |
|
|
|
|
|
-NO2 |
нитро |
- |
*Атом углерода, заключенный в скобки, входит в состав родоначальной структуры. **Алкокси-группы и все следующие за ними перечисляются в префиксе по алфавиту и не имеют
порядка старшинства.
Рациональная (радикально-функциональная) номенклатура
используется для названий простых моно- и бифункциональных соединений и некоторых классов природных соединений. Основу названия составляет название данного класса соединений или одного из членов гомологического ряда с указанием заместителей. В качестве локантов, как правило, используются греческие буквы.
3.2.3 Изомерия органических соединений Изомерия – это явление существования отличающихся по свойствам
химических соединений с одинаковым качественным и количественным составом и молекулярной массой, т. е. с одинаковой молекулярной формулой.
Типы изомерии: структурная и пространственная
Понятие «изомеры» введено Берцелиусом в 1830 г. Он определил «изомеры» как вещества, имеющие одинаковый состав (молекулярную формулу), но различные свойства. Представление об изомерах Берцелиус ввел после того как установил, что циановая кислота НОСN идентична по составу гремучей или изоциановой кислоте О=С=NН.
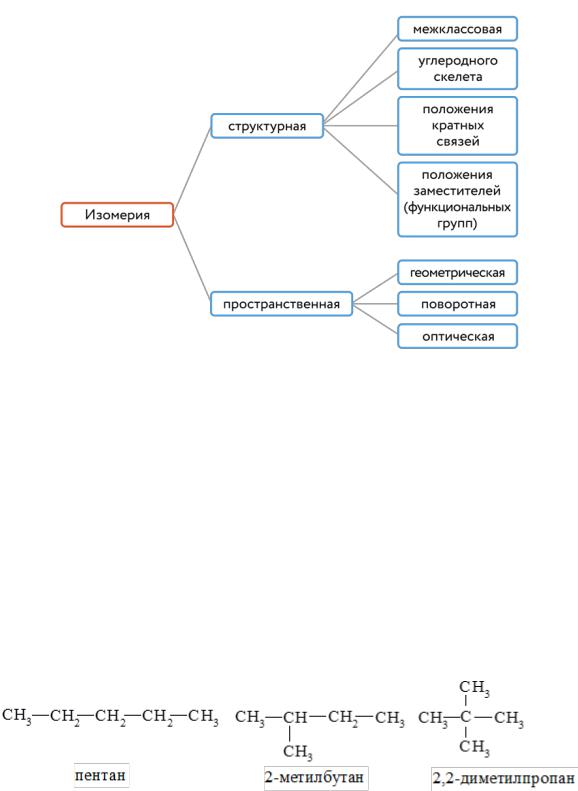
293
Различают два основных вида изомерии: структурную и пространственную (стереоизомерию) (рисунок 3.2.3.1).
Рисунок 3.2.3.1 – Виды изомерии органических соединений
Структурные изомеры отличаются друг от друга порядком связей между атомами в молекуле; стереоизомеры — расположением атомов в пространстве при одинаковом порядке связей между ними. Таким образом, структурная изомерия базируется на теории химического строения А.М. Бутлерова, а пространственная изомерия основывается на стереохимических представлениях Вант–Гоффа и Ле Беля.
Структурная изомерия подразделяется на несколько разновидностей (Рисунок 1).
Изомерия углеродного скелета обусловлена различным порядком связи между атомами углерода, образующими скелет молекулы.
Так, например, а) для предельных углеводородов:
б) для этиленовых углеводородов:
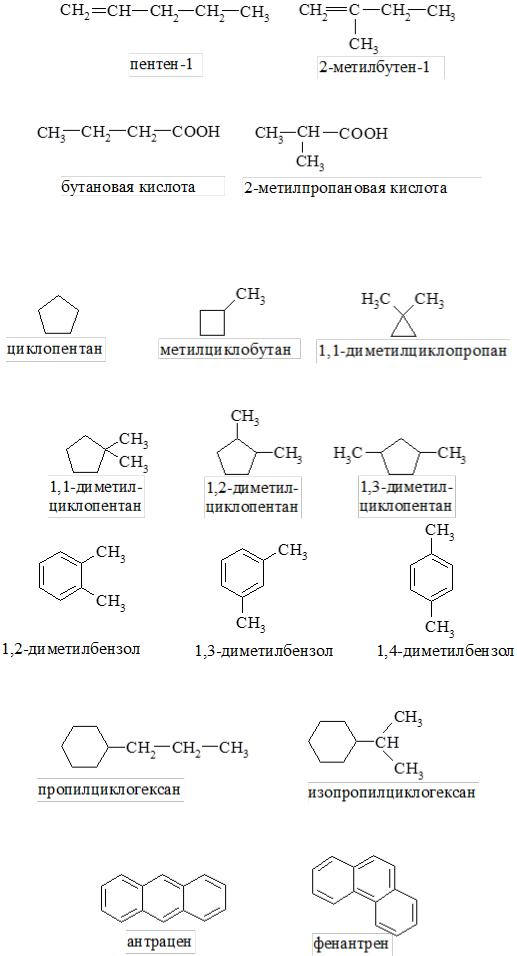
294
в) для карбоновых кислот:
В случае карбоциклических соединений к скелетной относятся следующие виды изомерии:
а) изомерия по величине цикла:
б) изомерия по положению углеродных заместителей в кольце:
в) изомерия боковых цепей:
г) изомерия по взаимному положению колец:
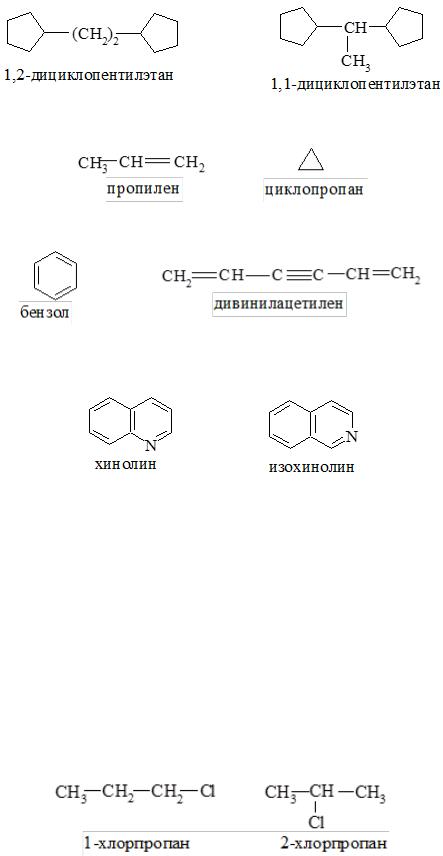
295
Примером цикло-цепной скелетной изомерии является изомерия алкенов и алициклов:
К этому же виду относятся изомерные бензол и дивинилацетилен:
В случае гетероциклов скелетными являются изомеры, отличающиеся
положением гетероатома в несимметричном кольце:
Особым видом структурной изомерии является таутомерия (равновесная динамическая изомерия) – существование вещества в двух или более изомерных формах, легко переходящих друг в друга. Например, ацетоуксусный эфир существует в виде равновесной смеси кетонной и енольной форм:
СН3С(ОН)=СНСООС2Н5 |
СН3С(О)СН2СООС2Н5 |
енольная форма |
кетонная форма |
Изомерия положения заместителей – при одном и том же углеродном скелете соединения могут отличаться лишь положением неуглеродного радикала, такие вещества называются изомерами по положению заместителя, например:
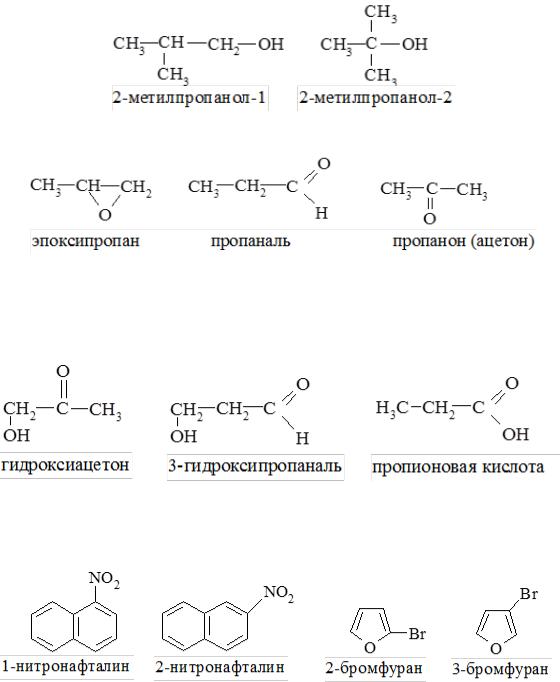
296
К данному виду следует отнести и изомерию альдегидов, кетонов и эпоксидов:
Если заместителей два или больше, общее число изомеров при прочих равных условиях возрастает, так как появляются изомеры по взаимному положению заместителей. Например, известно 4 структурных изомера формулы С4Н9Сl и 9 изомеров С4Н8Сl2. При различных заместителях изомеры могут относиться к разным классам органических соединений:
Существуют многочисленные примеры изомеров по положению заместителей для карбоциклических и гетероциклических соединений, например,
В ряду алифатических простых эфиров, сульфидов и аминов существует специальный вид изомерии – метамерия, обусловленная различным положением гетероатома в углеродной цепи. Метамерами являются, например, метилпропиловый и диэтиловый эфиры:
СН3–О–СН2–СН2–СН3 |
СН3– СН2–О–СН2–СН3 |
Изомерия положения кратных связей – обусловлена различием кратных связей, их положением в углеродном скелете или их взаимным расположением. Изомерами, например, являются ацетиленовые и диеновые углеводороды:
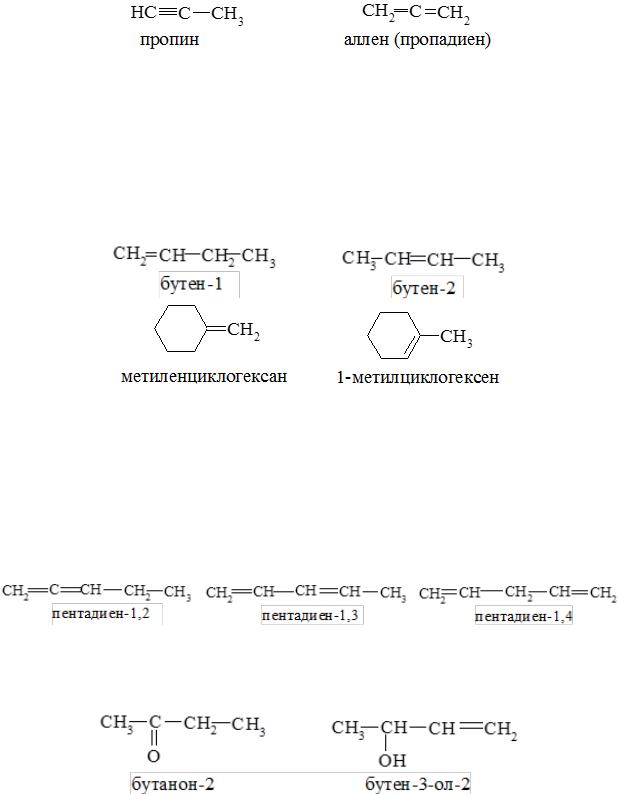
297
Такую же молекулярную формулу – СnH2n–2 имеют также циклоалкены и бициклические углеводороды, ибо каждая двойная связь, как и кольцо, приводит к уменьшению числа атомов водорода на 2. Следовательно, они также являются изомерами алкинов и алкадиенов, но изомерами скелетного типа.
Единственным отличием приведенных ниже изомеров является положение двойной связи:
В последнем примере двойная связь в одном случае находится вне цикла (экзоциклическая), в другом – внутри него (эндоциклическая). Как правило, эндо-соединение более устойчиво.
Известная классификация диенов на кумулированные, сопряженные и изолированные. Она основана на взаимном положении двух двойных связей. При одном и том же углеродном скелете – это изомеры по положению двойной связи:
К этому же виду относится изомерия альдегидов и кетонов с непредельными спиртами, например,
Межклассовая изомерия обусловлена различным положением и сочетанием атомов в молекулах веществ, имеющих одинаковую молекулярную формулу, но принадлежащих разным классам.
Итак, примеры изомерных классов органических веществ:
а) Алкены и циклоалканы, например, гексен–1 и циклогексан:
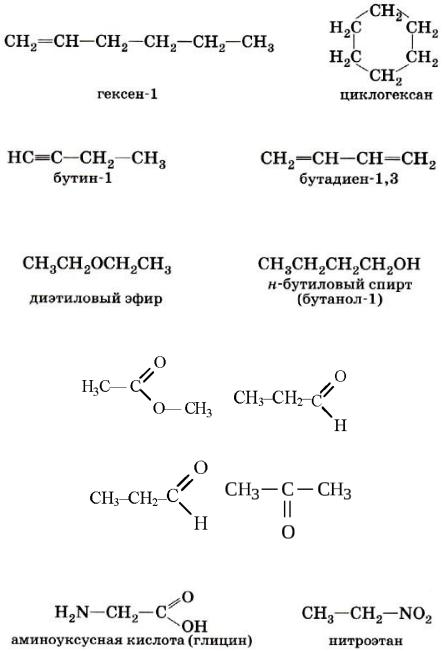
298
б) Алкины и алкадиены (диены), например, бутин–1 и бутаиен–1,3:
в) Простые эфиры и одноатомные спирты, например, диэтиловый эфир и бутанол–1:
г) Сложные эфиры и монокарбоновые кислоты, например, метилацетат и пропановая кислота:
д) Альдегиды и кетоны, например, пропаналь и пропанон–2:
е) Аминокислоты (содержащие один атом азота и одну карбоксильную группу) и нитроалканы, например, глицин и нитроэтан:
Пространственная изомерия подразделяется на два вида:
конфигурационную и конформационную. Конфигурационная в свою очередь делится на геометрическую изомерию (или цис-транс- изомерию) и
оптическую изомерию.
Геометрическая изомерия наблюдается в соединениях, содержащих жесткий фрагмент, т.е. двойную связь или цикл. Атомы или группы атомов могут располагаться по-разному относительно этого фрагмента. Полученное их расположение называется – конфигурация.
Для соединений с двойной связью возможность геометрической изомерии возникает при наличии двух неодинаковых заместителей у каждого из атомов, связанных двойной связью. Как двойная связь, так и цикл являются жесткими и препятствуют вращению атомов или групп атомов вокруг линии связи. Два заместителя могут располагаться по разные стороны
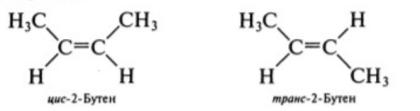
299
плоскости двойной связи или кольца или по одну сторону. В первом случае это – транс-изомеры, во втором – цис-изомеры.
Цис-изомеры – более полярные, более растворимы в полярных растворителях, более высококипящие, но низкоплавкие, менее устойчивые. Транс-изомеры – менее полярные, менее растворимые в полярных растворителях, более низкокипящие, но более высокоплавкие, более устойчивые.
На плоскости цис–, транст-изомеры изображают в виде проекционных формул. На примере молекулы 2-бутена это выглядит следующим образом:
Отличия в свойствах геометрических изомеров вызваны разными расстояниями между группами атомов возле двойной связи и различной степенью взаимодействия между соответствующими группами и атомами. Так, у транс-изомера заместители максимально удалены друг от друга и испытывают наименьшее взаимное отталкивание, тогда как в цис-изомере они сближены, и взаимодействие их выражено в наибольшей степени. Поэтому геометрические изомеры обладают разными физико–химическими свойствами, а в ряде случаев отличаются и некоторыми химическими реакциями. Превращение изомеров друг в друга при обычных условиях невозможно без разрыва двойной связи. Поэтому цис–транс-изомеры существуют как индивидуальные соединения.
Для названия геометрических изомеров используется также E, Z
номенклатура (расположение старших заместителей).
Например, цис-2-бутен |
транс-2-бутен |
Z-бутен-2 |
E-бутен-2 |
Z (zusammen-вместе) |
E (entgegen-напротив) |
Старшинство заместителей определяется атомным номером элемента, атом которого связан с атомом углерода двойной связи, а при одном и том же элементе – атомными номерами элементов, следующих по цепи заместителя.
Ряд заместителей в порядке возрастания старшинства.
Если относительное старшинство группы нельзя определить сравнением номеров первых атомов, то необходимо провести аналогичное сравнение для следующих атомов в группах (и так далее, если необходимо, двигаясь дальше от углерода двойной связи или цикла).
Оптическая изомерия была открыта еще задолго до теории химического строения, т.е. в начале 19 столетия. Было найдено, что при пропускании поляризованного света через некоторые вещества, они вращают (отклоняют) плоскость поляризации плоскополяризованного света на некоторый угол. Причем находятся всегда два изомера, которые отклоняют
300
на одну и ту же величину угла, но в разные стороны. Такая способность получила название оптической активности, а вещества, обладающие такими свойствами, стали называть оптически активными.
Начнем с того, что дело имеют с плоскополяризованным светом. В поляризованном свете поперечное колебание совершается только в одной плоскости, перпендикулярной направлению распространения светового луча. Плоскость поляризации – плоскость перпендикулярная к плоскости поперечных колебаний.
Явление оптической изомерии обнаруживают с помощью прибора поляриметра. На рисунке 3.2.3.2 приведена схема модифицированного поляриметра СМ-3.
Рисунок 3.2.3.2 – Блок-схема экспериментальной установки: 1 – источник излучения (светодиод), 2 – поляризатор, 3 – гнездо для кюветы с раствором оптически активного вещества, 4 – анализатор, 5 – шкала поворота поляризатора, 6 – телескопическая система
Явление вращения плоскости поляризации схематически показано на рисунке 3.2.3.3
Рисунок 3.2.3.3 – Явление вращения плоскости поляризации
Из рисунка видно, ориентация плоскости колебаний вектора →Е после прохождения оптически активного вещества изменяется на угол Dφ. В этом случае говорят, что произошел поворот плоскости поляризации на угол Dφ.
Главная причина проявления оптической активности – общая асимметрия молекулы.
Также асимметричные молекулы могут существовать в виде двух ассиметричных форм, которые при любом вращении не совмещаются друг с другом. В таких молекулах обычно присутствует асимметричный атом (хиральный центр), в частности асимметричный углеродный атом. Это такой атом, который имеет разные заместители, в случае углерода четыре разных заместителя.
301
Для такой молекулы можно построить две модели, которые асимметричны (не имеют элементов симметрии).
Пример: молочная кислота:
В молекуле имеется асимметрический атом углерода (С*), поэтому существуют два пространственных изомера молочной кислоты, являющихся зеркальными изомерами.
Два стереоизомера, относящиеся друг к другу как предмет и его зеркальное отражение, называют антиподами, или энантиомерами (см. рисунок 4).
Антиподы имеют одинаковый качественный и количественный состав, одинаковое химическое строение, идентичны по физико–химическим и химическим свойствам и отличаются только знаком оптического вращения.
Оптические изомеры отличаются друг от друга порядком расположения четырех разных заместителей вокруг асимметричного центра. Этот порядок расположения называется конфигурацией.
Вместо моделей, особенно когда число асимметричных центров больше единицы, пользуются проекционным формулами, которые впервые были предложены Э. Фишером – т.е. проекционные формулы Фишера.
Чтобы получить проекционные формулы, модель определенным образом ориентируем относительно плоскости проекции: главную цепочку расположим вертикально, самую окисленную функцию – наверху, а изгиб выпуклостью направляем на наблюдателя.
|
|
COOH |
|
|
||
H |
|
|
|
OH |
COOH |
|
|
|
|
|
|
||
|
|
|||||
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|||
|
|
|
|
|||
|
|
|
|
|||
|
|
|
|
H |
|
OH |
|
|
|
|
|
СН3 |
|
Теперь нужно решить очень важный вопрос: какому оптическому изомеру какая конфигурация у асимметричного центра соответствует?
Пошли по пути выбора стандартного соединения. Ими были выбраны глицериновые альдегиды. Право- и левовращающим глицериновым альдегидам приписаны нижеприведенные конфигурации.
|
|
CHO |
|
|
CHO |
|||||
H |
|
|
|
OH |
HO |
|
|
|
|
H |
|
|
|
|
|
|
|
||||
|
|
|
CH2OH |
|
|
|
|
CH2OH |
||
|
|
|
|
|
|
|||||
D (+) глицериновый альдегид |
L (-) глицериновый альдегид |
|||||||||
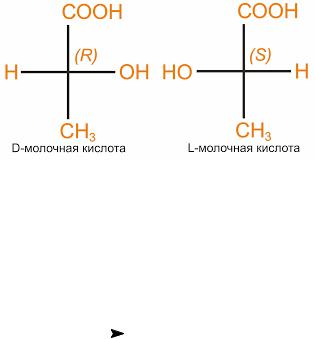
302
Для правовращающего глицеринового альдегида, применяли конфигурацию у асимметричного центра, которая изображена на левом рисунке. Такую конфигурацию обозначили большой буквой D. Конфигурацию левовращающего изомера обозначили буквой L.
Итак, имеются всего два стерических ряда D и L, и, сравнивая конфигурацию определенного (последнего) асимметричного углеродного атома соединения с конфигурацией глицериновых альдегидов относят его к одному из этих рядов.
Теперь, сравнивая конфигурации асимметрического центра у молочных кислот с конфигурацией стандарта можно отнести их к определенному стерическому ряду.
Теперь нужно решить какой кислоте соответствует какое вращение. Этот вопрос решается переходом от вещества с известным вращением и известной конфигурацией к данному веществу, например, к молочной кислоте. При этом нужно провести такие реакции, которые не затрагивают асимметричный центр.
|
|
CHO |
|
|
|
COOH |
||||
|
|
[ O] |
|
H |
|
|
OH |
|||
H |
|
|
|
OH |
|
|
|
|||
|
|
|
|
|
|
|
||||
|
|
|
[ H |
] |
|
|
||||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2OH |
|
|
|
CH3 |
||||
|
|
D (+) |
|
|
|
|
D (-) |
|||
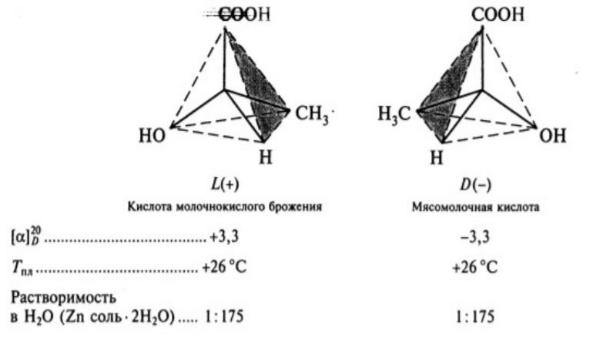
303
При этом из D – глицеринового альдегида получается D – молочная кислота, которая, согласно эксперименту на поляриметре, оказалась левовращающей.
Значит, левовращающаяся молочная кислота имеет D- конфигурацию. Молочная кислота, получающаяся при молочнокислом брожении, является
левовращающей (–), а выделяемая из мяса — правовращающей (+) (рисунок 3.2.3.4).
Рисунок 3.2.3.4. – Антиподы молочной кислоты и их свойства
Синтетическая молочная кислота не обнаруживает оптических свойств, так как представляет собой равномолекулярную смесь антиподов. Такую смесь называют рацематом. Процесс превращения одного энантиомера в другой называют
рацемизацией.
Как правило, самопроизвольная рацемизация не происходит при обычных условиях. Превращения антиподов возможны лишь в случае разрыва ковалентных связей при асимметрическом атоме углерода.
R, S номенклатура органических изомеров
Для точного обозначения расположения заместителей у асимметричного центра в пространстве (абсолютная конфигурация) применяется R, S номенклатура. Обозначение R или S приписывается конфигурациям асимметричного центра, исходя из старшинства заместителей. Поэтому, прежде всего определяется какие заместители у асимметричного центра и какой их порядок старшинства. Правила определения ряда старшинства те же, что и при использовании E, Z- номенклатуры геометрических изомеров. Далее каждый асимметрический центр молекулы ориентирует в пространстве таким образом, чтобы глаз наблюдателя смотрел по оси углерод – младший заместитель, чаще всего С–H. Определяем направление (порядок) расположения трех оставшихся заместителей от старшего к младшему: если по часовой стрелке, то конфигурация асимметричного атома обозначается префиксом R (rectus), если против часовой стрелки – префиксом S (sinister).
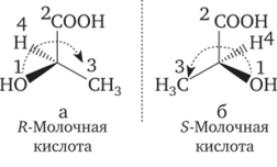
304
R |
S |
(по часовой стрелке) |
(против часовой стрелки) |
С ростом числа асимметричных углеродных атомов в молекуле увеличивается стереоизомеров. Если нет дополнительных элементов симметрии, то при числе асимметричных центров, равном n, количество оптических изомеров равна N= 2n, где n – число асимметричных центров.
Рассмотрим общий случай при n = 2 и N = 4.
Структуры I и II (см. ниже) по отношению к III и IV не относятся предмет к своему зеркальному изображению. Это не антиподы, не зеркальные изомеры, не энантиомеры.
Их называют диастереоизомерами или диастререомерами. Они различаются не только по отношению к плоскополяризованному свету, но и по физическим и химическим свойствам, т.к. расстояния между функциональными группами разные.
|
|
|
B |
|
B |
|
|
|
B |
|
|
B |
||||||||||
A |
|
|
|
|
C |
C |
|
|
A |
A |
|
|
|
C |
C |
|
|
|
|
A |
||
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
E |
|
|
D |
E |
|
|
|
D |
D |
|
|
|
|
E |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
D |
|
|
|
E |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
F |
|
|
|
F |
|
|
|
|
|
|
||||
|
|
|
F |
|
|
|
|
III |
|
|
F |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
II |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
I |
|
|
|
|
|
|
|
|
|
IV |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
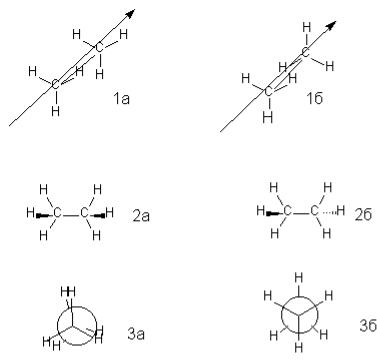
305
Конформации (поворотная изомерия) Переход от простейшего органического углеводорода – метана, к его ближайшему гомологу – этану ставит проблемы пространственного строения, для решения которых недостаточно знать рассмотренные в разделе параметры. В самом деле, не меняя ни валентных углов, ни длин связей, можно представить себе множество геометрических форм молекулы этана, отличающихся друг от друга взаимным поворотом углеродных тетраэдров вокруг соединяющей их связи С-С. В результате такого вращения возникают поворотные изомеры (конформеры). Энергия различных конформеров неодинакова, но энергетический барьер, разделяющий различные поворотные изомеры, для большинства органических соединений невелик. Поэтому при обычных условиях, как правило, нельзя зафиксировать молекулы в одной строго определенной конформации: обычно в равновесии сосуществуют несколько легко переходящих друг в друга поворотных форм.
Способы графического изображения конформаций и их номенклатура таковы (рисунок 3.2.3.5). Рассмотрение начнем с молекулы этана. Для нее можно предвидеть существоввание двух максимально различающихся по энергии конформаций. Они изображены ниже в виде перспективных проекций (1) («лесопильные козлы»), боковых проекций (2) и формул Ньюмена (3).
Рисунок 3.2.3.5 – Способы графического изображения конформаций этана
Вперспективной проекции (1а, 1б) связь С-С надо представить себе уходящей вдаль; стоящий слева углеродный атом приближен к наблюдателю, стоящий справа – удален от него.
Вбоковой проекции (2а, 2б) четыре Н-атома лежат в плоскости чертежа; атомы углерода на самом деле несколько выходят из этой
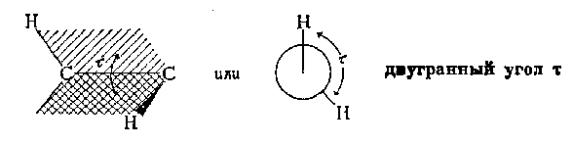
306
плоскости, но обычно упрощенно считают их также лежащими в плоскости чертежа. «Жирные» клиновидные связи утолщением клина показывают на выход из плоскости по направлению к наблюдателю того атома, к которому обращено утолщение. Пунктирные клиновидные связи отмечают удаление от наблюдателя.
В проекции Ньюмена (3а, 3б) молекулу рассматривают вдоль связи С-С (в направлении, указанном стрелкой на формулах 1а, б). Три линии, расходящиеся под углом 120 о из центра круга, обозначают связи ближайшего к наблюдателю углеродного атома; линии, «высовывающиеся» из-за круга – связи удаленного углеродного атома.
Изображенную слева конформацию называют заслоненной: название это напоминает о том, что атомы водорода обеих СН3-групп находятся друг против друга. Заслоненная конформация имеет повышенную внутреннюю энергию, и поэтому невыгодна. Конформацию, изображенную справа, называют заторможенной, подразумевая, что свободное вращение вокруг связи С-С «тормозится» в этом положении, т.е. молекула существует преимущественно в этой конформации.
Минимум энергии, необходимый для полного вращения молекулы вокруг определенной связи называется барьером вращения для данной связи. Барьер вращения в молекуле, подобной этану, может быть выражен через изменение потенциальной энергии молекулы как функции изменения двугранного (торсионного) угла системы. Двугранный угол (обозначаемый тау) изображен на рисунке 3.2.3.6:
Рисунок 3.2.3.6 – Изображение двугранного угла
Энергетический профиль вращения вокруг связи С-С в этане показан на рисунке 6. Вращение «заднего» атома углерода изображено изменением двугранного угла между двумя показанными атомами водорода. Для простоты остальные атомы водорода опущены. Барьер вращения, разделяющий две формы этана, составляет только 3 ккал/моль (12,6
кДж/моль). Минимумы кривой потенциальной энергии соответствуют заторможенным конформациям, максимумы – заслоненным.
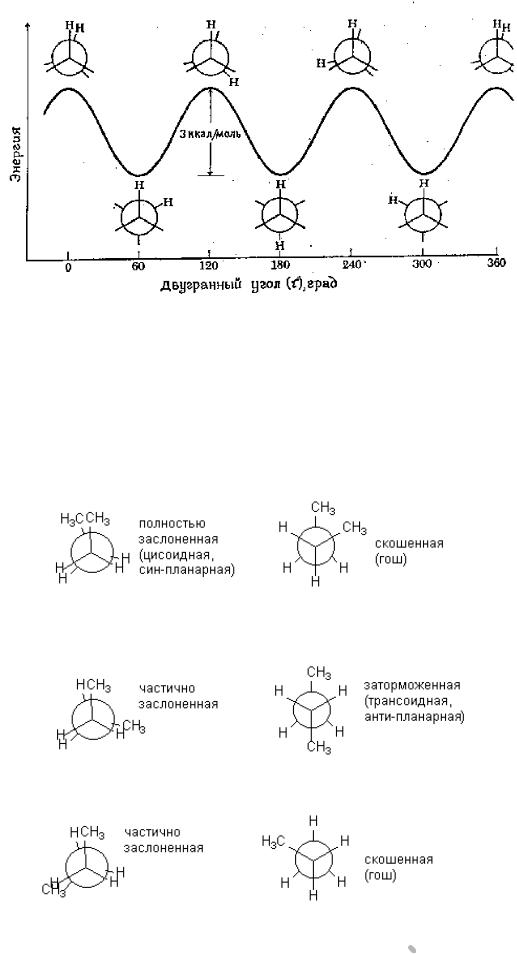
307
Рисунок 3.2.3.7 – Энергетический профиль вращения вокруг связи С-С в молекуле этана
С усложнением молекулы число возможных заметно отличающихся по энергии (характерных) конформаций возрастает. Так, для н-бутана при вращении вокруг связи С2-С3 можно изобразить уже шесть конформаций, отличающихся взаимным расположением СН3-групп, т.е. поворотом вокруг центральной связи С-С. Ниже конформации н-бутана изображены в виде проекций Ньюмена (рисунок 3.2.3.8).
Рисунок 3.2.3.8 – Конформации бутана
308
Изображенные слева (заслоненные) конформации энергетически невыгодны, практически реализуются лишь заторможенные.
Первая конформация наиболее неустойчивая (энергия 5,0 ккал/ моль относительно анти-конформера). В ней действует два типа напряжения:
торсионные и ван-дер-вальсовы и здесь они максимальны.
Торсионные напряжения возникают, когда взаимодействуют электроны противолежащих связей. В заслоненных конформациях они максимальны. Ван-дер-вальсовы напряжения возникают, когда атомы или группы атомов приближаются на расстояние, равное или меньше суммы их вандер- вальсовых радиусов.
В гош-конформации торсионные напряжения минимальны (заторможенная конформация), однако имеются довольно сильные ван-дер- вальсовые усилия (относительная энергия 0,9 ккал/моль)/ в третьей и пятой конфигурации действуют максимальные торсионные усилия и имеются еще ван-дер-вальсовые напряжения (относительная энергия 3,5 ккал/моль). Наконец, в анти-конформациях минимальны, как и торсионные, так и ван- дер-вальсовые усилия (энергия принята равной 0,0 кк/моль).
Итак, конформации – это различающиеся по внутренней энергии состояния молекулы, которые возникают при свободном вращении вокруг одинарной связи. Конформеры – это стереоизомерные структуры, находящиеся в подвижном равновесии и способные к взаимопревращению путем вращения вокруг простых связей.
309
3.3 ЭЛЕКТРОННОЕ СТРОЕНИЕ И РЕАКЦИОННАЯ СПОСОБНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПЛАН
3.3.1Строение органических соединений
3.3.2Кислоты и основания (Бренстед, Льюис).
3.3.3Электронные и пространственные эффекты в органических
реакциях.
3.3.1 Строение органических соединений
Типы связей в молекулах органических соединений, σ- и π- -связи. Гибридизация
Химическая связь – совокупность взаимодействий между электронами и ядрами, приводящих к соединению атомов в молекулу. Основные типы химической связи в органических соединениях – ионная,
ковалентная и водородная.
Ионная связь встречается в основном в неорганических соединениях. Она образуется при взаимодействии атомов, которые сильно отличаются по электроотрицательности. В этом случае происходит переход валентных электронов от атомов с меньшей электроотрицательностью к атомам, у которых она больше. В результате такого перехода возникают два противоположно заряженных иона – катион и анион, которые взаимно притягиваются друг к другу.
Для органических соединений наиболее характерными являются ковалентная и водородная связи.
Ковалентная связь
Ковалентная связь – связь возникает в результате обобществления валентных электронов, которые до образования связи принадлежали двум атомам, электроотрицательности которых равны или отличаются незначительно.
Характеристики ковалентной связи
•Длина связи – расстояние между центрами связанных атомов;
•Валентный угол – угол между двумя связями, имеющими общий
атом;
•Ковалентный радиус – половина длины ковалентной связи между
атомами;
•Энергия связи – энергия, выделяющаяся при образовании химической связи и характеризующая ее прочность (200 1000 кДж/моль);
•Энергия диссоциации – энергия, необходимая для гомолитического расщепления отдельной связи в молекуле (для двухатомных молекул равна энергии связи).
Свойства ковалентной связи:

310
•Направленность – связь атомов осуществляется в том направлении, в котором обеспечивается максимальное перекрывание орбиталей;
•Насыщаемость – способность атомов образовывать ограниченное число ковалентных связей;
•Полярность – результат неравномерного распределения электронной плотности. Существуют полярная и неполярная ковалентные связи. Если ковалентная связи образуется между атомами с одинаковой электроотрицательностью, то общая электронная пара находится на одинаковом расстоянии от ядер обоих атомов – такая связь называется неполярной, существует в молекулах простых веществ: H2, N2, Cl2, O2 и т.д. Связь же с неравномерным распределением электронной плотности между взаимодействующими атомами называется полярной.
•Дипольный момент связи (μ) – векторная величина,
характеризующая полярность связи.
Разновидности ковалентных связей;
Ковалентные связи, при образовании которых область перекрывания электронных облаков находится на линии, соединяющей ядра атомов, называются σ–связями.
π-связи – это ковалентные связи, при образовании которых область перекрывания электронных облаков находится по обе стороны от линии, соединяющей ядра атомов. π-Связи образуются в тех случаях, когда между двумя атомами возникают две или три общие электронные пары.
Водородная связь
Водородная связь является особым видом химический связи. В образовании этой связи участвует атом водорода, который в данной молекуле уже связан обычной ковалентной связью с атомом какого-либо элемента, имеющего большую электроотрицательность (например, с фтором, кислородом, азотом).
Водородная связь может быть не только межмолекулярной (рисунок 3.3.1.1), но и внутримолекулярной. Например, в салициловом альдегиде (рисунок 3.3.1.2) происходит образование водородной связи между атомом водорода группы –OH и атомом кислорода группы >C=O:
Рисунок 3.3.1.1 – |
Образование |
Рисунок 3.3.1.2 – |
Образование |
межмолекулярной |
водородной |
внутримолекулярной |
водородной |
связи |
|
связи |
|
311
Внутри- и межмолекулярные водородные связи существуют в молекулах белков, ДНК и др.
Таким образом, при образовании водородной связи атом водорода находится между двумя атомами с высокой ЭО: с одним из них он связан обычной ковалентной связью, а с другим – водородной связью.
При образовании ковалентной связи в молекулах органических соединений общая электронная пара заселяет связывающие молекулярные орбитали, имеющие более низкую энергию. В зависимости от формы МО (σ-МО или π- МО) образующиеся связи относят к σ- или π-типу.
σ–связь (Error! Reference source not found.3.3.1.3) – ковалентная связь,
образованная при перекрывании s-, p- и гибридных АО вдоль оси, соединяющей ядра связываемых атомов (т.е. при осевом перекрывании АО). Одинарные связи всегда являются σ–связями.
Рисунок 3.3.1.3 – Образование σ-связи
π-связь (рисунок 3.3.1.4) – ковалентная связь, возникающая при боковом перекрывании негибридных р-АО. Такое перекрывание происходит вне прямой, соединяющей ядра атомов. π-связи возникают между атомами, уже соединенными σ-связью (при этом образуются двойные и тройные ковалентные связи). π-связь слабее σ-связи из-за менее полного перекрывания р-АО.
Рисунок 3.3.1.4 – Образование π-связи между атомами углерода
Число общих электронных пар между связанными атомами характеризует кратность связи. Если связь между двумя атомами образована
312
двумя общими электронными парами, то такая связь называется двойной связью. Любая двойная связь состоит из одной σ связи и одной π связи.
Если связь между двумя атомами образована тремя общими электронными парами, то такая связь называется тройной связью. Любая тройная связь состоит из одной σ связи и двух π связей.
Различное строение σ и π молекулярных орбиталей определяет характерные особенности σ и π связей
1.σ Связь прочнее π связи. Это обусловлено более эффективным осевым перекрыванием АО при образовании σ МО и нахождением σ- электронов между ядрами;
2.Электроны на π МО, находясь вне межъядерного пространства, обладают большей подвижностью по сравнению с σ электронами. Поэтому поляризуемость π связи значительно выше, чем σ связи;
3.По σ-связям возможно внутримолекулярное вращение атомов, т.к. форма σ МО допускает такое вращение без разрыва связи;
4.В связи с отсутствием вращательной симметрии у π МО (из-за наличия узловой плоскости) поворот по двойной (σ + π) связи невозможен без разрыва π связи.
Гибридизация Гибридизация атомных орбиталей – изменение формы и энергии
орбиталей атома при образовании ковалентной связи для достижения более эффективного перекрывания орбиталей.
Основные положения теории гибридизации
1.Введение гибридных орбиталей служит для описания направленных локализованных связей. Гибридные орбитали обеспечивают максимальное перекрывание АО в направлении локализованных σ-связей;
2.Число гибридных орбиталей равно числу исходных АО, участвующих в гибридизации;
3.Гибридизуются близкие по энергии АО, независимо от того заполнены они в атоме полностью, наполовину или пусты;
4.В гибридизации участвуют АО, имеющие общие признаки
симметрии.
Гибридизация атомных орбиталей возможна лишь для атомов, образующих химические связи, но не для свободных атомов
sp3-гибридизация
Одна s- и три р-орбитали смешиваются, и образуются четыре равноценные по форме и энергии sp3-гибридные орбитали (рисунок 3.3.1.5). Оси sp3-гибридных орбиталей направлены к вершинам правильного тетраэдра. Тетраэдрический угол между ними равен 109°28', что соответствует наименьшей энергии отталкивания электронов. Характерна для алканов.
313
Рисунок 3.3.1.5 – Образование sp3-гибридных орбиталей
sp2-гибридизация
Происходит при смешивании одной s- и двух p-орбиталей (рисунок 3.3.1.6). Образуются три гибридные орбитали с осями, расположенными в одной плоскости и направленными к вершинам треугольника под углом 120°. Негибридная p-атомная орбиталь перпендикулярна плоскости и, как правило, участвует в образовании π-связей. Характерна для алкенов.
Рисунок 3.3.1.6 – Образование sp2-гибридных орбиталей
sp-гибридизация
Происходит при смешивании одной s- и одной p-орбиталей (рисунок 3.3.1.7). Образуются две равноценные sp-атомные орбитали, расположенные линейно под углом 180° и направленные в разные стороны от ядра центрального атома. Две оставшиеся негибридные p-орбитали располагаются во взаимно перпендикулярных плоскостях и участвуют в образовании π-связей, либо занимаются неподелёнными парами электронов. Характерна для алкинов.
Рисунок 3.3.1.7 – Образование sp-гибридных орбиталей
Длина связи с участием атома углерода зависит от его гибридизации: при увеличении кратности связи ее длина уменьшается; энергия связи уменьшается с увеличением вклада s-орбиталей в гибридизованное состояние: sp3 (25,0% s-АО) > sp2 (33,3% s-АО) > sp (50,0% s-АО); чем длиннее связь, тем меньше ее энергия (т.е. связь слабее). Характеристики

314
некоторых ковалентных связей представлены в таблице 3.3.1.
Таблица 3.3.1 – Характеристики ковалентных связей
Связь |
Гибридизация |
Энергия, |
Длина, нм |
Дипольный |
|
атома C |
кДж/моль |
момент, D |
|||
|
|
||||
|
sp3 |
348 |
0,154 |
0 |
|
|
sp2 |
626 |
0,134 |
0 |
|
|
sp |
814 |
0,120 |
0 |
|
|
|
|
|
|
|
|
sp3 |
414 |
0,110 |
0,30 |
|
|
sp2 |
435 |
0,107 |
0,40 |
|
|
sp3 |
344 |
0,143 |
0,86 |
|
|
sp2 |
708 |
0,121 |
2,40 |
|
|
sp3 |
293 |
0,147 |
0,45 |
|
|
sp2 |
598 |
0,128 |
1,40 |
|
|
sp3 |
451 |
0,140 |
1,39 |
|
|
sp3 |
331 |
0,176 |
1,47 |
|
|
|
460 |
0,096 |
1,51 |
|
|
|
|
|
|
|
|
|
390 |
0,101 |
1,31 |
|
|
|
|
|
|
|
|
|
348 |
0,130 |
0,70 |
|
|
|
|
|
|
Представление о механизмах реакций. Гомо- и гетеролитический разрыв связей. Представление о промежуточных частицах. Классификация реагентов
Классификация органических реакций
Существуют разные системы классификации органических реакций, которые основаны на различных признаках. Среди них можно выделить классификации:
По конечному результату реакции, то есть изменению в структуре субстрата (по структурному признаку);
По механизму протекания реакции, то есть по типу разрыва связей и типу реагентов.
По структурному признаку
В соответствии с этим признаком различают следующие типы реакций:
Реакция присоединения
,
например: |
. |
К реакциям присоединения относят также реакции полимеризации, в результате которых происходит последовательное присоединение молекул друг к другу за счёт разрыва кратных связей, например,

315
Этилен |
Полиэтилен |
Реакция замещения |
|
В органической химии к |
реакциям замещения могут относиться |
реакции, в которых один атом или функциональная группа замещается на другой атом или функциональную группу Y:
Однако, если в неорганической химии к реакциям замещения относят только реакции между простым и сложным веществами, то в органической химии в реакциях замещения могут участвовать одни только сложные вещества. Примерами реакций замещения с участием органических веществ могут служить следующие реакции:
(реакция между простым и сложным веществом)
(реакция, в которой все участвующие вещества сложные).
Реакция отщепления (элиминирования)
,
например:
Реакция изомеризации, например, изомеризация енолов в карбонильные соединения:
Окислительно-восстановительные реакции
Часто окисление сопровождается введением в молекулу органического вещества кислорода и (или) отщеплением водорода. Так, в результате окисления первичных спиртов образуются альдегиды, которые затем окисляются до карбоновых кислот:
За некоторыми типами реакций закрепились особые названия. Так, реакции присоединения водорода называют гидрированием, а реакции отщепления водорода – дегидрированием. Приставка «де» означает лишение
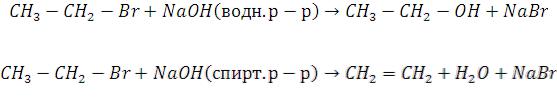
316
чего-либо. Аналогично, присоединение воды называют реакцией гидратации, а отщепление воды – реакцией дегидратации, присоединение галогеноводородов – гидрогалогенированием, а его отщепление — дегидрогалогенированием. Реакции, в результате которых в молекулу вводится галоген, называют реакциями галогенирования,  –группа –
–группа –
нитрования и т. д.
Реакции, в результате которых вещество подвергается термическому разложению без доступа воздуха, называются пиролизом. Примеры пиролиза
– сухая перегонка каменного угля, а также пиролиз метана.
Существуют также фотохимические реакции, протекающие в условиях освещения, например реакция фотосинтеза, которая происходит в зелёных растениях под действием света:
6СО2 + 6Н2О  С6Н12О6 + 6О2.
С6Н12О6 + 6О2.
В органической химии большую роль играют условия проведения реакций, в зависимости от которых могут получаться разные продукты с одними и теми же реагентами. Например, реакция бромэтана с водным раствором щёлочи приводит к образованию этанола, а со спиртовым — этена:
При составлении уравнений реакций важно указывать условия проведения реакций.
По типу разрыва химических связей в реагирующих частицах
Согласно этой классификации, в органической химии выделяют
радикальные и ионные реакции.
Взависимости от внешних условий и от энергии связи ковалентная химическая связь может разрываться двумя способами: гомолитически и гетеролитически.
Врезультате гомолитического (радикального) разрыва связи каждый атом получает неспаренный электрон и образуются две частицы, называемые
свободными радикалами. Например,  .
. – радикал.
– радикал.
Они представляют собой частицы, в которых атом углерода имеет три sp2-гибридизованные атомные орбитали, с помощью которых способен образовывать три ковалентные связи и один неспаренный электрон. Они могут иметь как плоское строение, подобно карбокатионам, так и пирамидальное, как карбанионы. В большинстве случаев радикалы существуют короткое время и присутствуют в малых количествах.
|
|
317 |
|
|
|
Гомолитический |
разрыв |
характерен |
для |
неполярных |
или |
слабополярных молекул и требует большого количества энергии, поэтому реакции с гомолитическим разрывом происходят при сильном нагревании или при облучении светом. Реакции, протекающие по радикальному механизму, характерны, например, для алканов.
Образующиеся радикалы, имеющие неспаренный электрон, обладают высокой реакционной способностью, поэтому химические процессы, протекающие с участием таких частиц, часто носят «цепной» характер, их трудно контролировать, а в результате реакции получается набор продуктов
замещения. Так, при хлорировании метана |
продуктами |
замещения |
|
являются хлорметан |
, дихлорметан |
, хлороформ |
и |
четырёххлористый углерод |
. |
|
|
Гетеролитически разрываются связи между атомами, электроотрицательность которых различается значительно. В результате гетеролитического (ионного) разрыва связи электронная пара остается у более электроотрицательного атома и образуются две заряженные частицы – ионы: катион (положительный) и анион (отрицательный). Гетеролитический разрыв характерен соединений, в которых существуют полярные ковалентные связи, в которых электронная пара смещена к более электроотрицательному атому.
Реакции с гетеролитическим разрывом связи протекают, как правило, в полярных растворителях (воде, безводной уксусной кислоте, хлороформе). Примером реакции, протекающей по ионному механизму, может служить реакция взаимодействия галогеналканов с водным раствором щелочи, например:  .
.
В результате гетеролитического разрыва связи образуются заряженные частицы: нуклеофильная (нуклеофил) и электрофильная (электрофил).
Нуклеофилы предоставляют электронную пару для образования новой
связи. |
Примерами нуклеофилов являются любые анионы, например |
, , |
и др., а также соединения, имеющие неподелённую электронную |
пару, например  ,
,  и др.
и др.
Электрофильные частицы с пониженной электронной плотностью на реакционном центре; содержат либо целый положительный заряд, либо электронодефицитный атом углерода или гетероатом:  и др. Для
и др. Для
образования ковалентной связи электрофил предоставляет свободные орбитали электронам частицы, с которой взаимодействует.
Частицу с положительным зарядом на атоме углерода называют карбокатионом. Карбокатионы, или карбениевые ионы представляют собой
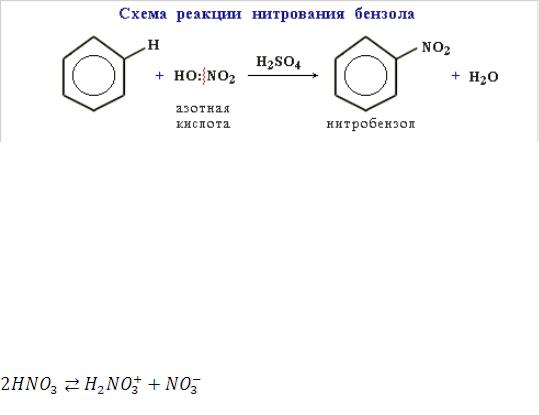
318
интермедиаты, у которых атом углерода имеет на внешнем уровне три электрона. Ковалентные связи, образованные за счет sp2-орбиталей расположены в одной плоскости под углом 120°С, поэтому, карбокатионы являются плоскими частицами. Их можно разделить на первичные, вторичные и третичные. Наиболее устойчивы те, у которых заряд находится на третичном атоме углерода.
Карбанионы представляют собой интермедиаты, у которых атом углерода имеет пять электронов. Ковалентные связи, образованные за счёт sp3-орбиталей расположены под углом близким по значению к 100°С, поэтому, карбоанионы имеют пирамидальную структуру.
Известны два основных метода получения карбанионов. Первый из них
– депротонирование С-Н-кислот. В зависимости от силы кислот депротонирование можно осуществить действием амминов, гидроксидов щелочных металлов в водных растворах или алкоголятов в спирте, амида натрия в жидком аммиаке. Чрезвычайно сильными основаниями являются металоорганические соединения, особенно литийорганические реагенты.
Классификация реагентов
Органическую реакцию рассматривают как взаимодействие органического соединения с реагентом.
Например, реакция нитрования:
Субстрат |
Реагент |
Продукт |
Субстрат – вещество, подвергающееся химическому превращению. Реагент – вещество, под действием которого субстрат подвергается
химическому превращению.
Как правило, химические превращения осуществляет не сам реагент, а атакующие частицы, которые из него образуются:

 +
+  +
+  O нитроний-катион
O нитроний-катион
нитрацидий-катион.
Энергетический профиль реакции; энергетический барьер реакции, энергия активации, тепловой эффект реакции. Кинетический и термодинамический контроль
Суть химической реакции состоит в превращении одного или нескольких химических веществ (исходные вещества), в одно или несколько
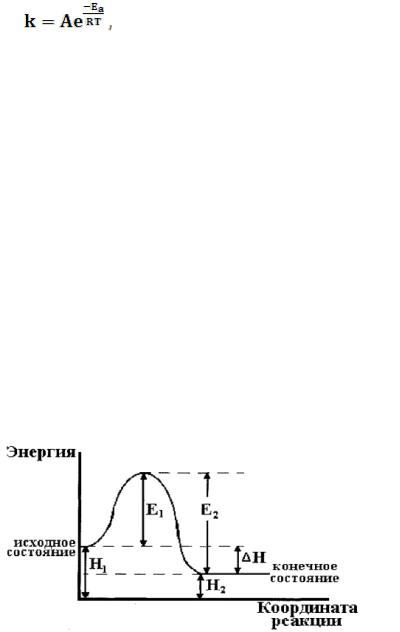
319
других химических веществ (продукты реакции). В большинстве случаев химическая реакция осуществляется не просто путем прямого перехода молекул исходных веществ в молекулы продуктов реакции, а состоит из нескольких стадий.
Совокупность стадий, из которых складывается химическая реакция,
называется механизмом или схемой химической реакции.
Множитель k, показывающий, с какой скоростью идет химический процесс при концентрациях реагирующих веществ, называется константой скорости химического процесса. Константа скорости химической реакции, как правило, резко растет с повышением температуры. Зависимость константы скорости реакции от температуры в большом числе случаев может быть описана уравнением Аррениуса (1):
(1)
Где, k – константа скорости,
R – универсальная газовая постоянная, T – температура,
Ea – энергия активации,
е – предэкспоненциальный коэффициент (учитывает вероятность и число столкновений).
Для простых реакций параметр Е показывает, какой минимальной энергией должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. Частицы, энергия которых ≥ Е, называются активными, а параметр Е в связи с этим называют энергией активации. Типичные значения Еа лежат в диапазоне от 10 до 100 кДж·моль-1 и по порядку величины близки к энергии химических связей.
Энергия активации связана с энтальпией (количеством теплоты, которое выделяется или поглощается системой в результате протекания химической реакции). И их связь иллюстрируется с помощью энергетических кривых химических реакций, или энергетического профиля реакции
(рисунок 3.3.1.8).
Рисунок 3.3.1.8 – Энергетический профиль экзотермической реакции

320
Однако, поскольку не все молекулы вступают в химическую реакцию, а только обладающие повышенным запасом энергии, то вводится понятие об
энергетическом барьере химической реакции, то есть необходимости исходным молекулам, прежде чем превратиться в продукты реакции, преодолеть активационный барьер. Для прямой химической реакции это будет энергия E1. Для обратной химической реакции это будет барьер Е2.
Прохождение молекулы через какой-либо энергетический барьер можно назвать путём реакции. Поскольку пути реакций могут быть различны, то и высота энергетических барьеров может изменяться. Значит, должна существовать точка, через которую будет проходить путь, имеющий самый низкий энергетический барьер. Геометрия молекулярной системы, соответствующая этой точке, и получила название переходного состояния или активированного комплекса. Комплекс похож на молекулу, но в отличие от нее не устойчив к распаду на продукты реакции. Если энергии столкновения оказалось достаточно для его образования, то реакция будет успешно завершена.
В 1930-х годах была разработана теория переходного состояния,
которую так же называют теорией активированного комплекса. Эта теория обосновала закон действия масс для элементарных реакций, то есть пропорциональность скорости реакций произведению концентраций участвующих в реакции частиц, и общий вид зависимости константы от температуры, а также позволила рассчитать для ряда реакций предэкспоненциальные множители в хорошем согласии с экспериментальными данными. Основные моменты, которые можно вынести из теории:
Взаимодействие исходных веществ не сразу приводит к образованию продуктов. Вначале образуется «переходное состояние» или активированный комплекс (рисунок 3.3.1.9).
Рисунок 3.3.1.9 – Образование активированного комплекса
Активированный комплекс – это неустойчивое соединение, в которое входят все атомы столкнувшихся и вступивших во взаимодействие молекул. Время жизни активированного комплекса мало.
Через некоторое время после образования активированный комплекс распадается с образованием молекул продуктов; при этом выделяется энергия.
321
Выделяющаяся при распаде активированного комплекса энергия может полностью или частично затрачиваться на активацию других молекул исходных веществ.
Количество выделенной (или поглощенной) теплоты называют тепловым эффектом реакции. Изучением тепловых эффектов химических процессов занимается раздел химии – термохимия. Уравнения реакций, в которых указаны тепловые эффекты, называются термохимическими.
Теплота образования (энтальпия образования) – это тепловой эффект реакции получения 1 моль сложного вещества из простых веществ, взятых в наиболее устойчивых модификациях при стандартных условиях.
Энтальпия образования простого вещества в стандартном состоянии равна нулю при любой температуре.
В ходе химической реакции протекают два процесса – разрыв химических связей в исходных веществах и образование новых связей в продуктах реакции. Разрыв химических связей всегда идёт с затратой энергии, сопровождается поглощением теплоты Q1 и является эндотермическим процессом. Образование новых химических связей – экзотермический процесс, протекающий с выделением теплоты Q2. Алгебраическая сумма тепловых эффектов этих стадий представляет собой
общий тепловой эффект реакции. Обозначается Q или H (Q = -ΔH),
измеряется в кДж или Дж.
Пример экзотермической реакции:
Пример эндотермической реакции:
Реакции образования устойчивых веществ из простых веществ экзотермические, реакции разложения чаще всего – эндотермические.
Кинетический и термодинамический контроль
Рассмотрим химическую реакцию, в которой участвуют два вещества А и В. В результате образуются продукты С и D (рисунок 3.3.1.10).

322
Рисунок 3.3.1.10 – Кинетический и термодинамический контроль
На данном рисунке видно, что образование продукта С происходит быстрее, так как он имеет меньшую энергию активации (Еа).
При низкой температуре, когда энергия реагирующих молекул невелика, основное направление реакции – образование соединения С. Соединение С образуется быстро. Его стабильность относительно низка, а энергия активации превращения продукта С в D невелика. Этот процесс контролируется скоростью и называется кинетически контролируемой реакцией, а соединение С – продуктом кинетического контроля.
Образование соединения D требует большей энергии, так как его Еа больше Еа продукта С, поэтому при низкой температуре оно – побочный продукт. Однако продукт D термодинамически более устойчив, чем С. Поэтому с ростом температуры, когда большее число молекул способно перебраться через высокий энергетический барьер, в реакционной массе наблюдается преобладание продукта D. Процесс образования соединения D контролируется энергетическими факторами и называется
термодинамически контролируемой реакцией. Соединение D – продукт
термодинамического контроля.
При проведении реакции при повышенной температуре (в условиях термодинамического контроля) происходит превращение соединения С в более энергетически выгодный продукт D.
Иначе говоря, термодинамический контроль – это необратимая и максимально выгодная химическая реакция. Продукты такого контроля всегда «стоят на первом месте» и их выход значительно больше. А вот кинетический контроль, наоборот, обратимая и менее энергетически выгодная химическая реакция, продукты которой «занимают второе место» и имеют меньший выход.
Рассмотрим эти два вида контроля на примере гидрогалогенирования диена:
1.При низких температурах
|
323 |
В результате выход продукта |
составил 20%, а |
выход продукта |
составил 80%. |
2.При повышении температуры
В результате выход продукта |
составил 80%, а |
выход продукта |
составил 20%. |
3.3.2 Кислоты и основания (Бренстед, Льюис).
Теория кислот и оснований Льюиса
Льюисом была предложена теория кислот и оснований, опирающаяся на строение внешних электронных оболочек атомов. По теории Льюиса кислотные и основные свойства соединений определяются их способностью принимать или отдавать электронную пару с образованием связи.
Основание Льюиса – это молекула (или анион), которая является донором электронной пары. Как правило, в образовании ковалентной связи с другой молекулой (или катионом) принимает участие несвязывающая (неподеленная) пара электронов основания. Другая молекула (или катион), с которой образует связь основание, является акцептором электронной пары и называется кислотой Льюиса.
Определив кислоты Льюиса как акцепторы электронной пары, можно прийти к выводу, что таких молекул должно существовать огромное множество. К ним должны относиться катионы металлов, органические катионы, нейтральные соли металлов (ZnCl2, AlCl3 и т.д.), соединения бора (BF3, BH3, R3B), металлоорганические соединения и т.п.
Большинство кислот Льюиса представляют собой многоэлектронные молекулы или катионы, однако протон, который также относится к кислотам Льюиса, не имеет ни одного электрона. Кроме того, протон очень мал по размерам и, следовательно, способен очень быстро двигаться, т.е. кислотноосновные реакции с участием протона часто могут идти очень быстро. Таким образом, протон среди кислот должен занимать особое место. Выделение протона из всех кислот Льюиса обусловлено также и его особой ролью в органической химии и биохимии.
Кислоты Льюиса можно разделить на классы в зависимости от того, с каким атомом кислоты координируется основание. Например, BF3 можно считать бор-кислотой (В-кислотой), поскольку реагирующее основание образует связь с бором. Когда трет-бутильный катион координируется с хлорид-ионом и дает трет-бутилхлорид, его можно считать С-кислотой. Ион нитрония (NO2+) в большинстве реакций действует как N-кислота и т.д.
Основаниями Льюиса являются молекулы, атомы или анионы, имеющие неподеленную пару электронов, которую они предоставляют для образования связи с вакантной орбиталью. К основаниям Льюиса относятся
324
спирты, простые эфиры, амины, тиоспирты, тиоэфиры, а также соединения, имеющие p-связи. В реакциях основания Льюиса проявляют себя как нуклеофильные частицы.
Экспериментальное развитие теории Льюиса привело к созданию принципа жестких и мягких кислот и оснований (Р. Пирсон).
По теории жестких и мягких кислот и оснований (ЖМКО) кислоты и основания Льюиса делятся на жесткие и мягкие.
Жесткие основания – это донорные частицы, обладающие высокой электроотрицательностью, низкой поляризуемостью, трудно окисляющиеся. Термин «жесткое основание» подчеркивает, что соединение прочно удерживает свои электроны. Донорными атомами в жестких основаниях могут быть кислород, азот, фтор, хлор.
Мягкие основания – это донорные частицы с низкой электроотрицательностью, высокой поляризуемостью, довольно легко окисляющиеся. Они слабо удерживают свои валентные электроны. В качестве доноров электронов выступают атомы углерода, серы, йода.
Жесткие кислоты – это кислоты Льюиса, в которых акцепторные атомы малы по размеру, и, следовательно, обладают высоким положительным зарядом, большой электроотрицательностью и низкой поляризуемостью. Низшая свободная молекулярная орбиталь жестких кислот, на которую переходят электроны донора, имеет низкую энергию.
Мягкие кислоты – это кислоты Льюиса, которые содержат акцепторные атомы большого размера, с малым положительным зарядом, с небольшой электроотрицательностью и высокой поляризуемостью. Низшая свободная молекулярная орбиталь этих соединений имеет высокую энергию. [3, с. 68].
Суть принципа ЖМКО состоит в том, что жесткие кислоты преимущественно реагируют с жесткими основаниями, а мягкие кислоты – с мягкими основаниями. Это выражается в большей скорости реакции и в образовании более устойчивых соединений, так как взаимодействие между орбиталями с близкими энергиями эффективнее, чем между орбиталями, имеющими разную энергию. Знание этого принципа полезно в качестве общетеоретической основы различных взаимодействий органических соединений.
Теория кислот и оснований Брёнстеда-Лоури
Кислоты Брёнстеда-Лоури – это молекулы или ионы, способные отдавать протон.
Основания Брёнстеда-Лоури – это молекулы или ионы, способные принимать протон.
Кислота, отдавая протон, превращается в основание, а основание, принимая протон, превращается в кислоту. Например, взаимодействие уксусной кислоты и воды протекает следующим образом - молекула воды отрывает протон от молекулы уксусной кислоты с образованием иона гидроксония и ацетат-иона:

|
325 |
|
СН3СООН + Н2О |
СН3СОО- + Н3О+ |
|
кислота основание |
сопряжённое |
сопряжённая |
|
основание |
кислота |
Следовательно, продуктами взаимодействия кислот и оснований Брёнстеда-Лоури являются кислоты и основания Брёнстеда-Лоури.
Для сравнения отметим, что продуктами взаимодействия кислот и оснований Аррениуса являются соли, в случае взаимодействия кислот и оснований Льюиса образуются молекулярные комплексы (таблица 3.3.2).
Таблица 3.3.2 – Характеристика теорий кислот и оснований
Авторы теории |
Основание |
Кислота |
|
|
|
|
|
|
Вещество, диссоциирующее в воде с |
Вещество, диссоциирующее в |
|
Аррениус |
образованием гидроксид-ионов |
воде с образованием протонов |
|
|
(NaOH, КОН) |
(H2SO4, HNO3, CH3COOH) |
|
Брёнстед-Лоури |
Акцептор протона |
Донор протона |
|
(NH3, H2O, амины, анионы и др.) |
(HCl, CH3COOH, H2O и др.) |
||
|
|||
|
Донор электронов |
Акцептор электронов |
|
Льюис |
(NH3, H2O, амины, анионы, |
(H+, AICl3, BF3, бензохинон, |
|
|
алкены, бензол и др.) |
тетрацианоэтилен и др.) |
Факторы, влияющие на силу кислоты
Сила кислоты количественно выражается через константу диссоциации: чем больше константа диссоциации, тем кислота сильнее.
Чем больше делокализован отрицательный заряд в сопряжённом основании, тем сильнее кислота. Факторы, приводящие к делокализации отрицательного заряда и поляризации связи А-Н приводят к увеличению силы кислоты:
1.Чем больше электроотрицательность (ЭО) атома, с которым связан протон, тем больше кислотность. Зависимость действует для кислотных центров – атомов одного периода.
Электроотрицательность: C < N < O < F Кислотность: CH4 < NH3 < H2O < HF.
2.Чем больше поляризуемость атома кислотного центра, тем больше кислотность.
Поляризуемость – это способность поляризоваться под действием внешнего электрического поля. Чем больше электронов в атоме и чем дальше они от ядра, тем больше поляризуемость. Поэтому кислотность водородных соединений элементов одной группы таблицы Менделеева, как правило, увеличивается с увеличением порядкового номера (сверху вниз). Например, кислотность в ряду галогеноводородных кислот увеличивается в следующем порядке: HF < HCI < HBr < HI
Порядок увеличения ЭО является противоположным. Можно сделать вывод, что поляризуемость здесь является более весомым фактором, чем ЭО.
Порядок изменения кислотности в случае водородных соединений

326
шестого периода - кислорода и серы аналогичен: H2O < H2S.
3. Электроноакцепторные заместители увеличивают кислотность, электронодонорные уменьшают.
ЭА увеличивают делокализацию отрицательного заряда в сопряжённом основании, увеличивают его стабильность и, следовательно, увеличивают силу кислоты.
Рассмотрим некоторые конкретные случаи.
Вопрос: какое соединение является более сильной кислотой – этанол (С2Н5ОН) или этилмеркаптан (С2Н5SH)?
Сера является более поляризуемой, чем кислород, поэтому этилмеркаптан является более сильной кислотой.
Вопрос: какое соединение является более сильной кислотой – уксусная кислота (СН3СООН) или хлоруксусная (СН2СlСООН)?
Атом хлора является сильным электроноакцептором. Электроноакцепторные заместители повышают силу кислоты. Следовательно, хлоруксусная кислота сильнее, чем уксусная.
Факторы, влияющие на основность
Факторы, увеличивающие основность, противоположны факторам увеличивающим кислотность. Чем меньше делокализована неподелённая электронная пара (НЭП) на основании и чем больший отрицательный заряд локализован на атоме – основном центре, тем более выражены основные свойства:
1)Чем больше электроотрицательность атома основного центра, тем сильнее удерживается НЭП, тем меньше основность. Зависимость действует для оснований, имеющих основные центры представленные атомами одного периода таблицы Менделеева.
Электрострицательность: N < О < F
Основность: NH3 > H2O > HF, т.е. аммониевые основания сильнее, чем оксониевые. Амины являются более сильными основаниями, чем спирты.
2)Чем больше поляризуемость атома основного центра, тем меньше основность.
Порядок основности: H2O > H2S , то есть оксониевые основания сильнее, чем сульфониевые.
3)Электронодонорные заместители увеличивают основность, электроноакцепторные уменьшают.
Рассмотрим некоторые конкретные случаи.
Вопрос: какое соединение является более сильным основанием: метиламин (СН3–NH2) или метанол (CH3–OH)?
Атомы кислорода и азота имеют неподеленную электронную пару, следовательно, они являются основными центрами. Атом кислорода является более электроотрицателен, чем атом азота, следовательно, метиламин является более сильным основанием, чем метанол.
Вопрос: какое соединение является более сильным основанием: метиламин (СН3–NH2) или диметиламин (CH3–NН–CH3)?
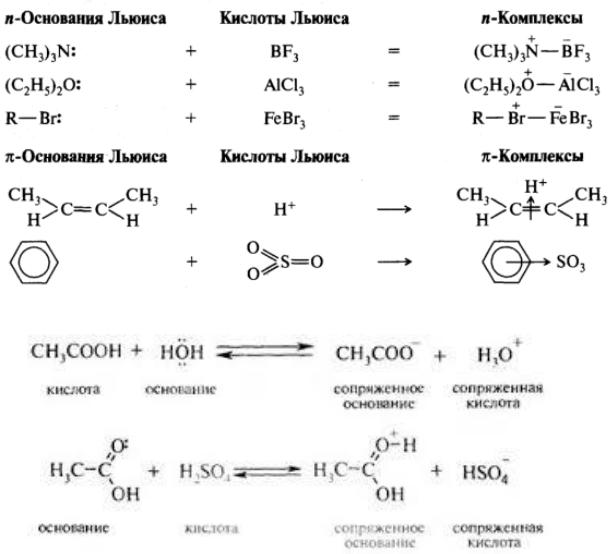
327
Основным центром является атом азота. В диметиламине две электронодонорные метильные группы, в то время как в метиламине только одна. Следовательно, диметиламин является более сильным основанием, чем метиламин.
Примеры взаимодействия кислот и оснований По Льюису:
По Брёнстеду-Лоури:
Константа кислотной ионизации и ее показатель (рКа).
Большинство органических соединений, проявляющих кислотные свойства, в водной среде являются слабыми кислотами, константы которых выражаются малыми величинами. Например, Кa уксусной кислоты при 25°С равна 1,76·10-5. Оперировать такими малыми величинами в практической работе не удобно, поэтому используют значения рКa – показатель константы
– отрицательный логарифм константы диссоциации (2):
рКa = – lgKa |
(2) |
Например, рКa для СН3СООН равняется 4,75.
Рассматривая зависимость между значениями величин Кa, рКa и силой кислоты, следует отметить одну закономерность: чем больше величина Кa

328
(константа кислотности), тем сильнее кислота, чем больше величина рКa, тем кислота слабее.
Подобно кислотам силу оснований иногда выражают величиной Кb (константа основности), характеризующей легкость, с которой основание отрывает протон от воды:
Kb
R–NH2 + HOH  R–NH3+ + OH-
R–NH3+ + OH-
Kb = [RNH3+] * [OH-] [R–NH2]
Для удобства пользуются величиной рКb (3):
pKb = -lg Kb |
(3) |
При этом, чем меньше рКb, тем сильнее соответствующее основание. Однако намного удобнее выражать степень ионизации кислот и оснований в одной шкале (в шкале рКb) подобно тому, как значение рН одинаково хорошо характеризует и кислотность, и основность. Для основания величина рКa обычно означает кислотность сопряженной кислоты – протонной формы основания рК+. Сопряженная кислота в виде R–NH3 отдавая протон, превращается в основание R–NH4+.
3.3.3 Электронные и пространственные эффекты в органических реакциях
Взаимное влияние атомов в молекулах, ионах, радикалах
В развитии органической химии важнейшую роль сыграло основное
положение классической теории А.М. Бутлерова: свойства вещества зависят не только от природы и числа входящих в него атомов, но и от химического строения молекул и взаимного влияния атомов.
Все составляющие молекулу атомы находятся во взаимосвязи и испытывают взаимное влияние. Это влияние передается в основном через систему ковалентных связей с помощью так называемых электронных эффектов: индуктивный эффект, эффект поля, мезомерный эффект,
гиперконьюгация.
Индуктивный эффект – смещение электронной плотности по цепи σ- связей, которое обусловлено различиями в электроотрицательностях атомов.
Одним из свойств ковалентной связи является некоторая подвижность электронной плотности. Она способна смещаться в сторону одного из атомов.
Если ковалентной связью связаны два совершенно тождественных атома, то электронная плотность будет равномерно распределена между этими атомами и связь будет неполярной. Атомы, связанные неполярной

329
ковалентной связью, не несут зарядов, они электронейтральны, например, в молекуле этана СH3–СН3.
Если один из атомов более электроотрицателен, чем его партнер по связи, то электронная плотность будет смещена в сторону этого атома и связь будет полярной.
Метанол Метан Хлорметан
Атомы, связанные полярной связью, несут частичные заряды, обозначаемые греческой буквой "дельта" (δ). Атом, оттягивающий электронную плотность σ-связи в свою сторону, приобретает частичный отрицательный заряд δ-. При рассмотрении пары атомов, связанных ковалентной связью, более электроотрицательный атом называют электроноакцептором. Его партнер по σ-связи соответственно будет иметь равный по величине дефицит электронной плотности, т.е. частичный положительный заряд δ+, и будет называться электронодонором. Например, если в неполярную молекулу метана СH4 ввести электроноакцепторные заместители, то их связь с атомом углерода будет полярной. Смещение электронной плотности полярной σ-связи обозначается прямой стрелкой, совпадающей с валентной черточкой.
Наличие полярной связи в молекуле сказывается на состоянии соседних связей. Они испытывают влияние полярной связи, и их электронная плотность также смещается в сторону электроотрицательного элемента, т.е. происходит передача электронного эффекта.
Смещение электронной плотности по цепи σ-связей называется
индуктивным эффектом и обозначается буквой I.
Индуктивный эффект передается по цепи с затуханием. Направление смещения электронной плотности всех σ-связей также обозначают прямыми стрелками.
Электроноакцепторные заместители, т.е. атом или группа атомов,
смещающие электронную плотность по σ-связи от атома углерода, проявляют отрицательный индуктивный эффект (-I-эффект). Электронодонорные заместители – атом или группа атомов, смещающие электронную плотность к атому углерода, проявляют положительный индуктивный эффект (+I- эффект).
+I-Эффект проявляют алифатические углеводородные радикалы, т.е. алкильные радикалы (метил, этил и т. д.).
330
Большинство функциональных групп проявляют -I-эффект: галогены, аминогруппа, гидроксильная, карбонильная, карбоксильная группы.
Эффект поля – действует не через связи, а непосредственно через пространство или молекулы растворителя. Часто очень трудно разделить эти два эффекта, но во многих случаях это было сделано; при этом, как правило, исходили из того факта, что эффект поля зависит от геометрии молекулы, а индуктивный эффект зависит только от природы связей.
pKa=5.67 |
pKa=6.07 |
Например, в показанных изомерах индуктивные эффекты атомов хлора, влияющие на положение электронов в группе СООН (следовательно, и на кислотность), одинаковы, поскольку между Сl и СООН находятся одни и те же связи, а эффекты поля различны, так как пространственное расположение в первом изомере таково, что атомы хлора находятся ближе к группе СООН, чем во втором изомере.
Полученные экспериментальные данные подтвердили, что во многих случаях эффекты поля гораздо более важны, чем индуктивные эффекты. Чаще всего оба типа эффектов рассматривают вместе.
Мезомерный эффект (эффект сопряжения) – смещение электронной плотности по цепи делокализованных (сопряженных) π-связей.
Многие соединения можно адекватно описать структурной формулой, однако для целого ряда других соединений этого недостаточно. Речь идет о соединениях, в которых одна или более связывающих орбиталей принадлежат не только паре атомов, но охватывают три ядра или даже большее число ядер.
Такого рода связь называют делокализованной. Поэтому используемый, обычно, химиками способ изображения связей между атомами в виде одной, двух или трех черточек, обозначающих участие в этих связях
Такого рода связь называют делокализованной. Поэтому используемый, обычно, химиками способ изображения связей между атомами в виде одной, двух или трех черточек, обозначающих участие в этих связях соответственно двух, четырех или шести электронов, совершенно неудовлетворителен. В действительности в образовании некоторых связей участвуют другие, и даже дробные числа электронов. Это можно видеть на примере этаноат-аниона (ацетат-анион) (рисунок 3.3.2.1), в котором, по данным рентгеноструктурного анализа, атомы кислорода неразличимы, а обе
331
углерод-кислородные связи имеют одинаковые межатомные расстояния, т.е. содержат одинаковое число электронов.
Рисунок 3.3.2.1 – Образование связей в этаноат-анионе
Эти трудности привели к тому, что молекулы, структуры которых не могут быть описаны в виде одной классической формулы, стали изображать комбинацией двух или большего числа классических структур, так называемых канонических структур, связанных стрелками с двумя остриями. Путь, по которому одна из этих структур может переходить в другую, часто указывают с помощью изогнутой стрелки, «хвост» которой показывает, откуда движется электронная пара, а ее острие – куда она движется.
Если группа С=O сопряжена со связью С=С, поляризация может передаваться дальше посредством π-электронов, как, например, для 2- бутеналь:
При этом происходит делокализация заряда, что приводит к нехватке электронов на атомах С3 и С1, как в простом карбонильном соединении. Различие между такой передачей через сопряженную систему и индуктивным эффектом в насыщенной системе состоит в том, что в данном случае при передаче по цепи эффект ослабляется в гораздо меньшей степени, а полярность соседних атомов углерода чередуется.
Направление смещения электронной плотности под влиянием М- эффекта обозначается изогнутыми стрелками.
+М-эффектом обладают заместители, повышающие электронную плотность в сопряженной системе. К ним относятся группы, которые содержат атомы с неподеленной парой электронов, способные к передаче этой пары электронов в общую систему сопряжения. +М-эффект характерен для групп -OH и -NH2. Так, в молекуле фенола C6H5OH группа -OH проявляет +М-эффект за счет участия одной из неподеленных электронных пар атома кислорода в системе сопряжения (рисунок 3.3.2.2).

332
Рисунок 3.3.2.2 – Проявление мезомерного эффекта в молекуле фенола
-М-эффект проявляют заместители с электроотрицательными атомами и смещающие электронную плотность на себя. -М-эффект характерен для групп - CH=O, -COOH, -NO2 (рисунок 3.3.2.3).
Рисунок 3.3.2.3 – Проявление мезомерного эффекта в молекуле бензальдегида
Хотя эти группы имеют неподеленные электронные пары, пространственное расположение орбиталей с этими электронами не позволяет им вступать в систему сопряжения. Таким образом, в данном случае заместитель может лишь оттягивать электроны из общей системы сопряжения за счет своей более высокой электроотрицательности.
Мезомерные эффекты, подобно индуктивным, вызывают поляризацию молекул в их основных состояниях и поэтому сказываются на физических свойствах соединений. Существенное различие между индуктивными и мезомерными эффектами состоит в том, что если индуктивные эффекты могут действовать как в насыщенных, так и в ненасыщенных соединениях, то мезомерные эффекты могут действовать только в ненасыщенных и,
333
особенно, в сопряженных соединениях. Индуктивные эффекты передаются только на сравнительно короткие расстояния в насыщенной цепи, в то время как мезомерные эффекты могут передаваться от одного конца сравнительно больших молекул к другому при условии наличия сопряжения (т. е. делокализованных π-орбиталей).
Сверхсопряжение (гиперконъюгация) – это взаимодействие σ-вязей с
π - системой. Это понятие распространяется на карбониевые ионы и радикалы, где имеется взаимодействие между σ-связями и незаполненными или частично заполненными р-орбиталями. Индуктивный эффект алкильных групп, как и следовало ожидать, уменьшается в ряду:
Однако если алкильные группы связаны с ненасыщенной системой, например, с двойной связью или бензольным кольцом, этот порядок нарушается и в случае некоторых сопряженных систем изменяется на обратный. Таким образом, по-видимому, алкильные группы способны при некоторых обстоятельствах вызывать смещение электронов с помощью механизма, отличного от индуктивного эффекта. Это удалось объяснить, расширив понятие сопряжения или мезомерного эффекта и предположив, что делокализация электронов происходит с участием электронов ζ-связей с π - системой.
Этот эффект получил название сверхсопряжения (гиперконъюгации), с
его помощью удалось объяснить ряд непонятных ранее явлений. Следует подчеркнуть, что в действительности не происходит отщепление протона или, поскольку, если он переместится из первоначального положения, то одно из условий, необходимых для делокализации, будет нарушено.
Обращение ожидаемого (индуктивного) ряда электронодонорных свойств (СНз > МеСН2 > Ме2СН > Ме3С) можно объяснить тем, что эффект гиперконъюгации зависит от наличия атомов водорода, связанных с углеродным атомом ненасыщенной системы. Ясно, что эффект гиперконъюгации должен быть максимальным у СН3-группы и отсутствовать у Ме3С-группы.
Эффект гиперконъюгации был использован для объяснения большей термодинамической устойчивости таких алкенов, у метилбутене-2, по сравнению с изомерными соединениями с концевой двойной связью,
334
например 2-метилбутене-1; в 2- метилбутене-2 имеется девять водородных атомов, способных участвовать в гиперконъюгации, а в 2-метилбутене-1 только пять:
2-метилбутен-2 2-метилбутен-1
В результате этого преимущественно образуются алкены с неконцевой двойной связью в реакциях, в которых возможно также образование соответствующих изомеров с концевой двойной связью, а также наблюдается очень быстрая изомеризация менее стабильного соединения в более стабильное (2-метилбутен-1 в 2-метилбутен-2).