


614
В кислой среде молекулы аминокислот представляю собой катион. В щелочной среде молекулы аминокислот представляют собой анион. В нейтральной среде аминокислоты представляют собой цвиттер-ион или
биполярный ион.
Аминокислоты в твердом состоянии всегда существуют в виде биполярного, двухзарядного иона — цвиттер-иона.
Водные растворы аминокислот в кислой и щелочной среде проводят электрический ток.
1. Взаимодействие внутри молекулы – образование внутренних солей (биполярных ионов)
Молекулы аминокислот существуют в виде внутренних солей, которые образуются за счет переноса протона от карбоксила к аминогруппе.
Карбоксильная группа аминокислоты отщепляет ион водорода, который затем присоединяется к аминогруппе той же молекулы по месту неподеленной электронной пары азота. В результате действие функциональных групп нейтрализуется, образуется так называемая внутренняя соль.
Водные растворы аминокислот в зависимости от количества функциональных групп имеют нейтральную, кислую или щелочную среду.
а) моноаминомонокарбоновые кислоты (нейтральные кислоты)
Внутримолекулярная нейтрализация — образуется биполярный цвиттер-ион.

615
Водные растворы моноаминомонокарбоновых кислот нейтральны
(рН≈7).
б) моноаминодикарбоновые кислоты (кислые аминокислоты)
Водные растворы моноаминодикарбоновых кислот имеют рН<7 (кислая среда), так как в результате образования внутренних солей этих кислот в растворе появляется избыток ионов водорода Н+.
в) диаминомонокарбоновые кислоты (основные аминокислоты)
Водные растворы диаминомонокарбоновых кислот имеют рН>7 (щелочная среда), так как в результате образования внутренних солей этих кислот в растворе появляется избыток гидроксид-ионов ОН— .
2. Взаимодействие с основаниями и кислотами
Аминокислоты как амфотерные соединения образуют соли как с кислотами (по группе NH2), так и со щелочами (по группе СООН).
Как кислота (участвует карбоксильная группа)
Как карбоновые кислоты α-аминокислоты образуют функциональные производные: соли, сложные эфиры, амиды.
а) взаимодействие с основаниями

616
Образуются соли:
б) взаимодействие со спиртами (р. этерификации)
Аминокислоты могут реагировать со спиртами в присутствии газообразного хлороводорода, превращаясь в сложный эфир. Сложные эфиры аминокислот не имеют биполярной структуры и являются летучими соединениями.
в) взаимодействие с аммиаком
Образуются амиды:
Как основание (участвует аминогруппа)
а) взаимодействие с сильными кислотами
Подобно аминам, аминокислоты реагируют с сильными кислотами с образованием солей аммония:
б) взаимодействие с азотистой кислотой (р. дезаминирования)
Подобно первичным аминам, аминокислоты реагируют с азотистой кислотой, при этом аминогруппа превращается в гидроксогруппу, а аминокислота – в гидроксикислоту:
Измерение объёма выделившегося азота позволяет определить количество аминокислоты (метод Ван-Слайка).

617
3. Внутримолекулярное взаимодействие функциональных групп
ε-аминокапроновой кислоты, в результате которого образуется ε-капролактам (полупродукт для получения капрона).
3. Межмолекулярное взаимодействие α-аминокислот – образование пептидов (р. поликонденсации):
4.
При взаимодействии карбоксильной группы одной молекулы аминокислоты и аминогруппы другой молекулы аминокислоты образуются пептиды. При взаимодействии двух α-аминокислот образуется дипептид.
Межмолекулярная реакция с участием трех α-аминокислот приводит к образованию трипептида и т.д.
Важнейшие природные полимеры – белки (протеины) – относятся к полипептидам, т.е представляют собой продукт поликонденсации α- аминокислот.
5. Качественные реакции! а) нингидриновая реакция
Все аминокислоты окисляются нингидрином с образованием продуктов
сине-фиолетового цвета:
Иминокислота пролин дает с нингидрином желтое окрашивание.
б) биуретовая реакция с ионами тяжелых металлов α-аминокислоты образуют внутрикомплексные соли. Комплексы меди (II), имеющие яркосинюю окраску, используются для обнаружения α-аминокислот.

618
в) ксантопротеиновая реакция используется для обнаружения а- аминокислот, содержащих в радикале ароматический цикл, например тирозина
При действии концентрированной азотной кислоты на раствор белка образуется нитросоединение, окрашенное в желтый цвет. При добавлении к нему щелочи окраска становится оранжевой в связи с ионизацией фенольной гидроксильной группы.
Представление о первичной структуре белков Под первичной структурой белков понимают последовательность
аминокислот в полипептидной цепи.
Первым исследователем, определившим аминокислотную последовательность молекулы белка, был Фред Сэнгер, работавший в Кавендишской лаборатории Кембриджского университета. Он работал с гормоном инсулином — самым маленьким белком, какой ему удалось найти. Работа заняла 10 лет и результаты ее были опубликованы в 1953 г. В 1958 г. Сэнгер за эту работу был удостоен Нобелевской премии (вторую Нобелевскую примию он получил за изучение структуры нуклеиновых кислот). В молекулу инсулина входит 51 аминокислота. Молекула состоит из двух полипептидных цепей, удерживаемых вместе дисульфидными мостиками.
Внастоящее время большая часть работ по определению аминокислотных последовательностей автоматизирована, и теперь первичная структура белков известна уже более чем для сотни тысяч белков.
Ворганизме человека тысячи различных белков, и все они построены из одних и тех же 20 стандартных аминокислот. Аминокислотная последовательность белка определяет его биологическую функцию. В свою очередь эта аминокислотная последовательность определяется нуклеотидной последовательностью ДНК. Замена одной-единственной аминокислоты в

619
молекуле данного белка может резко изменить его функцию, как это наблюдается, например, при так называемой серповидноклеточной анемии. Интересные данные могут быть получены в результате анализа аминокислотных последовательностей гомологичных белков, принадлежащих разным биологическим видам; такие данные позволяют судить о возможном таксономическом родстве между этими видами.
Синтетические полиамиды
Впервые синтетические полиамиды были получены в 1862 г. (поли-ц- бензамид) и в 1899 г. (поли-е-капрамид), а их промышленное производство было налажено в 1938 г. в США.
Из синтетических полиамидов практическое значение имеют алифатические и ароматические полиамиды. Их структура, конечно, отличается от структуры полипептидов, потому что амидные группы расположены дальше друг от друга в цепи и чередуются по направлению.
Гидролитическая деструкция белков и синтетических полиамидов протекает по амидной (пептидной) связи и катализируется щелочами и кислотами. Химическая структура природных белков близка к структуре синтетических полиамидов. Конечными продуктами гидролиза белков являются различные а-аминокислоты, синтетические полиамиды гидролизуются: с образованием исходных аминокислот или соответствующие дикарбоновых кислот и диаминов.
Из числа синтетических волокон капрон является наиболее широко известным. Синтезируется оно из 6-аминокапроновой кислоты:
Молекулы этой кислоты, имея па концах функциональные труппы с противоположными свойствами – основную и кислотную, вступают между собой в реакцию поликонденсации:
Такой процесс осуществляют в автоклаве при температуре около 250° С. В результате образуется высокомолекулярная смола – капрон. Молекулы капрона имеют линейное строение и содержат до 200 элементарных звеньев:

620
Как и в полипептидах, остатки аминокапроновой кислоты соединены между собой амидными связями:
Поэтому волокна из капрона относятся к группе так называемых
полиамидных волокон.
Наличие амидных связей роднит эти волокна с природными белковыми волокнами – шерстью и шелком. Полиамидные волокна, как и белковые, обладают высокой механической прочностью; в этом отношении они даже значительно превосходят природные.
Капроновое волокно, как и многие другие синтетические волокна, не впитывает влагу, не гниет, не поедается молью. Оно очень устойчиво к истиранию и к действию многократных деформаций, в чем превосходит все натуральные волокна.
Подобно белковым веществам, капрон недостаточно устойчив к действию кислот: по пептидной связи в нем происходит гидролиз. Сравнительно невысока и теплостойкость капронового волокна: при нагревании прочность его снижается, а при 215° С происходит плавление (поэтому изделия из капрона не рекомендуется гладить горячим утюгом). По светостойкости капроновое волокно уступает нитрону.
Известно, какое широкое применение находит капроновое волокно. Нарядные кофточки, шарфы, носки, чулки и многие другие изделия из капрона стали уже обычными в нашем быту. Большой популярностью пользуются изделия из витого капронового волокна – безразмерные, легко растягивающиеся чулки и носки. В последнее время из капрона стали готовить превосходные меховые изделия.
Капрон идет также на изготовление парашютных тканей, канатов, рыболовных снастей, лесок и т. д. Из упрочненного капрона делают кордную ткань, используемую в качестве каркаса авто- и авиапокрышек. Срок службы шин с кордом из капрона значительно выше срока службы шин с вискозным и хлопчатобумажным кордом.
Найлон (нейлон) — семейство синтетических полиамидов, используемых преимущественно в производстве волокон.
Наиболее распространены два вида нейлона: полигексаметиленадипинамид (анид (СССР/Россия), найлон 66 (США)), часто называемый собственно нейлоном, поли-ε-капроамид (капрон (СССР/Россия), найлон 6 (США)). Известны также другие виды, например: поли-ω-энантоамид (энант (СССР/Россия), найлон 7 (США)), поли-ω-
621
ундеканамид (ундекан (СССР/Россия), найлон 11 (США), рильсан (Франция, Италия)).
Полиамидное волокно анид (нейлон) получают из продукта совместной поликонденсации гексаметилендиамина H2N–(СН2)6–NH2 и адипиновой кислоты НООС–(СН2)4–СООН:
В промышленности нейлон применяется для изготовления втулок, вкладышей, пленок и тонких покрытий, струн. Он имеет низкий коэффициент трения и низкую температуру на трущихся поверхностях. Нейлон не растворяется в большинстве органических растворителей, не поддаётся воздействию слабых растворов кислот, щелочей и солёной воды.

622
3.8 ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
ПЛАН
3.8.1Классификация и номенклатура гетероциклов.
3.8.2Пятичленные гетероциклы.
3.8.3Шестичленные гетероциклы.
3.8.1 Классификация и номенклатура гетероциклов
Гетероциклические соединения – циклические соединения, в состав которых кроме атомов углерода и водорода входят другие, так называемые
гетероатомы – N, O, S, P.
пиррол |
фуран |
тиофен |
пирролин |
пирролидин |
имидазол |
тиазол |
оксазол |
пиридин |
пиримидин |
Наиболее важными гетероциклическими соединениями являются гетероциклы, содержащих атомы N, O, S (пятичленные, шестичленные и некоторые конденсированные гетероциклы). Они входят в состав многих веществ природного происхождения, таких как нуклеиновые кислоты, хлорофилл, алкалоиды, пенициллины, многие витамины. Гетероциклические соединения играют важную роль в процессах метаболизма, обладают высокой биологической активностью. Значительная часть современных лекарственных веществ содержит в своей структуре гетероциклы
Для классификации гетероциклических соединений используют следующие признаки:
-по размеру цикла гетероциклические соединения бывают чаще всего трех-, четырех-, пяти-, шести- и семичленными:
-природа гетероатома (O, S, N, P, Si, Bi, Te и др.):

623
-количество гетероатомов (моно-, диили три- и так далее гетероатомные циклы):
-степень насыщенности цикла (насыщенные, ненасыщенные и ароматические гетероциклы:
- в особую группу выделены конденсированные системы:
Номенклатура гетероциклов
Для гетероциклических соединений применяют тривиальные и систематические названия.
При построении систематических названий учитывается природа и количество гетероатомов, а также размер цикла и степень его насыщенности. Природа отражается в префиксе, размер цикла — в корне, а степень насыщенности — в суффиксе названия.

624
Для обозначения гетероатомов используют префиксы окса- (O), тиа-
(S), аза- (N) и др.
Размер цикла обозначается корнями -ир- (трех-), -ет- (четырех-), -ол- (пяти-), -ин- (шести-), -еп- (семичленный)
А степень насыщенности — суффиксами -идин (насыщенный цикл с атомом азота), -ан (насыщенный цикл без атома азота), -ин (ненасыщенный цикл).
В названии гетероциклов с максимально возможным количеством двойных связей в цикле суффикс не указывается.
Для частично гидрированных соединений используют приставки дигидро-, тетрагидро- с указанием номеров атомов, к которым присоединен водород.
Если атом водорода присоединен только к одному атому цикла, то в названии указывается номер гидрированного атома и символ Н.
В шести- и семичленных азотсодержащих гетероциклах полная насыщенность цикла обозначается приставкой пергидро-.
Количество гетероатомов одного вида указывают в названии множительными приставками ди-, три-, тетра- и т. д.
Если гетероцикл содержит несколько разных гетероатомов, то называют их в определенной последовательности: окса-, тиа-, аза- и др.
Нумерацию атомов в гетероцикле обычно начинают с гетероатома и проводят в том направлении, чтобы заместители получили меньшие номера.
В пяти- и шестичленных гетероциклах с одним гетероатомом атомы углерода иногда обозначают греческими буквами α, β, γ. Примеры:


626
CH=CH— замещена на гетероатом. Важнейшими представителями этой группы гетероциклов являются пиррол, фуран, тиофен:
Особенности строения
Согласно методу молекулярных орбиталей, молекулы пиррола, фурана, тиофена могут быть описаны как плоские пентагональные системы с sp2 – гибридизованными атомами углерода. Каждый атом кольца (как углерод так и гетероатом) связан σ-связями с тремя другими атомами. Для образования этих связей атом использует три sp2-орбитали, которые лежат в плоскости под углом 120 о.Таким образом, каждый атом затрачивает одни электрон на образование σ-связи, после чего у каждого атома углерода кольца остается один электрон, а у гетероатома – два электрона. Эти электроны занимают р- орбитали, которые, перекрываясь, образуют π -облако выше и ниже плоскости цикла. Так как эти π -облака содержат а сумме шесть электронов, образуется стабильная электронная оболочка («ароматический секстет»), которая придаёт стабильность циклу.
Структуры фурана, пиррола и тиофена аналогичны: отличие лишь в том, что если азот в пирроле несет атом водорода, то кислород или сера содержат неподеленную пару электронов на sр2-орбитали.
Индол содержит плоскую циклическую сопряженную систему, включающую 10 p-электронов, в том числе неподеленную пару электронов атома азота:
Таким образом, индол может быть отнесен к ароматическим p- избыточным гетероциклическим соединениям. Атом азота выступает как электронодонор, повышая электронную плотность на атомах углерода.
Физические свойства
Фуран, тиофен и пиррол – бесцветные жидкости, практически не растворимые в воде. Температуры их кипения значительно выше, чем у соответствующих им по числу углеродных атомов соединений жирного ряда,

627
а дипольные моменты ниже. Спектральные характеристики близки к характеристикам соединений ряда бензола.
Индол и его гомологи – бесцветные кристаллические вещества с неприятным запахом.
Особенности протекания реакций электрофильного замещения
Являясь ароматическими системами, пятичленные гетероциклы вступают преимущественно в реакции электрофильного замещения в специальных условиях. Их реакционная способность значительно выше реакционной способности бензола, для реакций требуются более мягкие реагенты. Примерный ряд изменения реакционной способности коррелируются с электроотрицательностью гетероатома (чем выше эта величина, тем выше реакционная способность гетероцикла):
бензол<тиофен<фуран<пиррол.
Пиррол
Для пиррола характерны реакции электрофильного замещения, SEAr.
1. Кислотные свойства (свойства NH-кислоты)
Пирролат калия используют для получения 1- и 2-алкилпирролов по следующей схеме:
2. Сульфирование пиррола проводят с помощью пиридинсульфотриоксида (серную кислоту нельзя использовать). На первом этапе образуется соль 2-пирролсульфокислоты и пиридина, который является третичным амином. Далее действуют более сильным основанием –

628
гидроксидом бария и получают бариевую соль 2-пирролсульфокислоты. Сульфокислоты пиррольного ряда нестабильны и их выделяют в виде бариевых солей:
3. Галоидирование
Для моногалоидирования используют диоксан-бромид и хлористый сульфурил, эти же реагенты применяют для моногалоидирования фенола. Бромирование бромом приводит к тетрабромпроизводному.

629
4. Формилирование, ацетилирование Формилирование – введение формильной группы (СНО) и
ацетилирование относятся к реакциям ацилирования. Они позволяют ввести карбонильную группу, которую можно легко модифицировать.
а) Реакция Вильмейера-Хаака В этой реакции в качестве донора формильной группы используют
диметилформамид:
б) Ацетилирование:
5. Гидрирование пиррола
Биологические свойства производных пиррола
Пиррол является родоначальником обширного класса соединений, относящихся к порфиринам, – это гемоглобин, хлорофилл, билирубин (красящее вещество желчи). В живой клетке эти пигменты синтезируются из порфобилиногена, участвующего в процессе основного метаболизма.
Получение

630
Фуран
Фуран вступает в реакции электрофильного замещения SEAr преимущественно по α-положению (аналогично пирролу).
1.Нитрование
4.Сульфирование
Сульфокислоты фуранового ряда также неустойчивы, как и кислоты ряда пиррола, поэтому их выделяют в виде солей.
3. Галоидирование
Галоидирование хлором протекает через промежуточное соединение - продукт 1,4-присоединения хлора с дальнейшим отщеплением 2-х молей хлористого водорода.
631
4. Формилирование, ацетилирование
Формилирование фурана проводится с помощью цианистого водорода в присутствии хлористого водорода, образующийся промежуточный имин гидролизуют:
Гидрирование
При гидрировании фурана получают насыщенный кислородсодержащий гетероцикл – тетагидрофуран, соединение относится к простым циклическим эфирам и используется в качестве растворителя в органическом синтезе.
Биологически активные производные фурана
Фурациллин – кристаллическое вещество ярко желтого цвета, ограниченно растворимое в горячей воде, обладает бактерицидными свойствами. Существует ряд производных нитрофурана, обладающих выраженными бактерицидными свойствами (фурадонин, фуразидин (фурогин, фуромаг)).
Фуросемид является диуретиком (снижает артериальное давление, уменьшает отеки). Применяется при лечении сердечно-сосудистых заболеваний, цирроза печени, отека легких, гипертонии.

632
Методы получения
Тиофен
Тиофен вступает в реакции электрофильного замещения SEAr. Он наименее реакционноспособен, по сравнению с пирролом и фураном, не ацидофобен.
1.Нитрование
2.Галоидирование
3.Сульфирование

633
4. Гидрирование
При гидрировании тиофена образуется насыщенный серусодержащий гетероцикл – тиофан, он является циклическим сульфидом. Этот гетероцикл входит в состав витамина Н.
Биологически активные производные тиофена Типепидин. Соединение обладает тиреоидным действием
(биорегулирующее влияние на щитовидную железу).
Эпросартан. Соединение обладает антигипертензивным действием (лечение гипертонии).
Пирантел, в виде тартрата – соли D-винной кислоты по вторичному и третичному атомам азота тетрагидропиримидинового цикла, обладает антигельминтным действием.
Методы получения
Индол
Все реакции электрофильного замещения в индоле идут по пиррольному кольцу. Существенным отличием от пиррола является ориентация электрофильного замещения в положение 3, что обусловлено более эффективной стабилизацией промежуточно образующегося катиона:

634
Имидазол и тиазол. Кислотно-основные свойства имидазола
Среди гетероциклических соединений наиболее многочисленна и разнообразна группа пятичленных гетероциклов, содержащих более одного гетероатома. Большинство этих циклических систем можно формально получить из фурана, пиррола и тиофена заменой одной или более групп — СН на гетероатом азота. Возможность варьирования числа и расположения атомов азота в кольце приводит к структурному многообразию гетероциклов.
Имидазол
Имидазол – более сильное основание, чем пиррол и пиразол, что проявляется в образовании межмолекулярных водородных связей, а, следовательно, и в более высокой температуре его кипения по сравнению с пиразолом:
имидазол |
пиразол |
пиррол |
Водородные связи имидазола:
Имидазол обладает амфотерностью, образует соли как с сильными кислотами, так и со щелочными и щелочноземельными металлами.
Реакции электрофильного замещения протекают либо по атомам азота, либо по атомам углерода, но наиболее предпочтителен первый вариант. Такой механизм реакции требует наименьшие энергетические затраты.
1. Алкилирование и ацилирование по атому азота
А) При нагревании имидазола с алкилирующими агентам (алкилгалогенидами или алкилсульфатами):
|
|
|
|
|
ÑH3 |
|
|
|
|
ÑH3 |
|
|
N |
|
|
|
N |
-H+ |
|
|
|
N |
|
|
|
|
|
|
|
|
|||||
|
|
+ |
|
|
|
||||||
|
+ CH3Br |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|||
N |
|
|
N Br- |
|
|
|
N |
||||
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
N-ì åòè ëè ì è äàçî ëè é |
|
1-м етилим идазо л |
|||||||
|
|
-áðî ì è ä |
|
|
|
|
|
|
|||
Б) Действие на имидазол галогенангидридов или ангидридов кислот приводит к образованию высокореакционноспособных N-ацильных производных.

|
|
635 |
|
|
|
|
N |
|
|
|
N |
|
|
||||
|
+ (CH3CO)2O |
|
|
|
+ CH3COOH |
|
|
||||
N |
|
|
N |
||
H |
|
|
|
|
|
|
|
|
|
ÑOCH3 |
|
|
|
|
N-ацети ли м и дазо л |
||
2. Замещение по атомам углерода
А) По отношению к электрофильным реагентам имидазол занимает промежуточное положение между пиридином и реакционноспособными пятичленными гетероциклами с одним гетероатомом (пирролом, фураном и тиофеном). По легкости вступать в реакции нитрования и сульфирования имидазол уступает и бензолу.
|
|
|
HNO (êî í ö), 1% î ëåóì , 200Ñ |
|
N |
|
|
|
||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|||
|
3 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
N |
NO2 |
|
|
N |
|
|
H |
|
|||||
|
|
|||||||||
N |
|
|
|
N |
|
5-н итро им идазо л, 90% |
||||
|
|
|
|
|
|
|
|
|
||
H |
î ëåóì , 1600Ñ |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
N |
|
SO3H |
|
||
H
им идазо л-5-сульф о н о вая кисло та, 60%
Б) В щелочной среде имидазолы со свободной NH-группой вступают в реакцию азосочетания:
N |
|
|
|
N |
|
|
+ ArN |
+ |
- |
pH >7 |
|
|
|
|
X |
|
|
|
|
|
2 |
|
N |
N |
N |
Ar |
|
N |
|
|
||||
H |
|
|
H |
|
|
|
В) Галогенирование имидазола в нейтральной среде, когда молекула неионизирована, протекает очень легко. Бромирование на холоду не останавливается на стадии монозамещения, а приводит к образованию тризамещенного продукта:
|
|
|
|
Br |
|
|
|
N |
Br CHCl , 00C |
|
N |
||
|
|
|||||
|
|
|
|
|||
|
|
2 |
3 |
|
|
|
N |
|
|
Br |
N |
Br |
|
|
|
|
||||
H |
|
|
|
H |
|
|

637
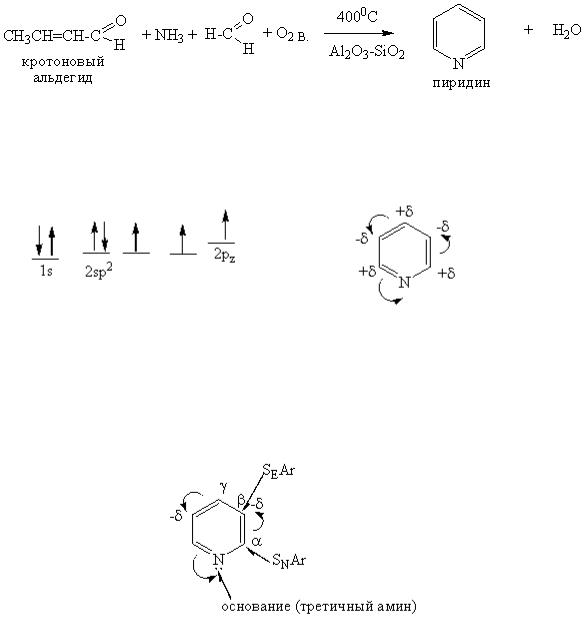
Пиридин
Пиридин – бесцветная жидкость с сильным неприятным запахом. При стоянии окисляется, желтеет.
Синтез пиридина
1)Из каменноугольной смолы;
2)По реакции Чичибабина:
Строение пиридина
Пиридин относится к ароматическим соединениям. Распределение электронов в атоме азота пиридина:
За счет большей электроотрицательности атома азота по сравнению с атомом углерода, электронная плотность в пиридине стянута к атому азота, поэтому ароматическая система пиридина называется – дефицитной.
Реакционная способность пиридина
Пиридин проявляет свойства третичного амина, вступает в реакции электрофильного замещения и реакции нуклеофильного замещения.
Пиридин относится к очень слабым основаниям. Причиной является электронное строение атома азота, который находится достаточно близко к ядру, что снижает возможности атома азота предоставлять пару электронов при образовании солей и проявлении нуклеофильных свойств. Тем не менее пиридин легко образует соли с минеральными и органическими кислотами, алкилируется и ацилируется.

638
1. Образование солей, алкилирование и ацилирование
2. Реакции электрофильного замещения
Реакции электрофильного замещения идут с трудом в очень жестких условиях:
3. Реакции нуклеофильного замещения
Реакции нуклеофильного замещения не были характерны для бензола и его гомологов. Введение в ароматическое ядро атома азота приводит к его обеднению электронами и, как следствие, появлению реакций нуклеофильного замещения. Но электороноакцепторные свойства атома азота недостаточно велики, поэтому реакции идут в жестких условиях.

639
4. Восстановление (гидрирование)
При восстановлении пиридина в зависимости от природы восстановителя получают циклические вторичные амины, содержащие в 3-ем положении двойную связь – пиперидеины-3, либо полностью насыщенные – пиперидины.
Пиперидин входит в состав природных соединений, например – алкалоиды конин и анабазин.
Биологически активные производные пиридина и пиперидина
1. Гомологи пиридина, пиридинкарбоновые кислоты и их производные.


642
Биологически активные производные хинолина
Хинозол, нитроксилин, энтеросептол являются антисептиками при инфекционных заболеваниях желудочно-кишечного тракта.
Изохинолин
Изохинолин – бесцветные кристаллы со слабым запахом миндаля; плохо растворим в холодной воде, в органических растворителях — хорошо.
Он содержится в небольшом количестве в каменноугольном дёгте, откуда его выделяют вместе с хинолином.
Изохинолин – более сильное основание, чем хинолин. Обладает однотипными химическими свойствами как хинолин.

643
При гидрировании над платиной превращается в 1,2,3,4- тетрагидроизохинолин; при полном гидрировании — в цисдекагидроизохинолин.
Окисление смесью озона и кислорода приводит к пиридин-3,4- дикарбоновой кислоте (цинхомероновая кислота), окисление пероксокислотами ведёт к изохинолин-N-оксиду.
Реакции электрофильного замещения происходят в положении 5.
Бромирование легче протекает в присутствии AlCl3, нитрование идёт при действии серной и азотной кислот. Сульфирование при действии 40% олеума при температуре 180оС приводит обычно к изохинолин-8-сульфокислоте. Сульфирование 60% серной кислотой при 300оС ведёт к смеси изохинолин-5- и изохинолин-8-сульфокислот.
При высокой температуре изохинолин вступает в реакции бромирования по радикальному механизму с образованием 1- бромизохинолина.
Нуклеофильное замещение протекает как правило в положение 1. С гидроксидом калия при 200оС изохинолин образует 1-гидроксиизохинолин, с амидом натрия – 1-аминоизохинолин.
Изохинолин является токсичным высокоопасным веществом по степени воздействия на организм.
Представление о природных азотсодержащих гетероциклических соединениях (алкалоидах, компонентах нуклеиновых кислот) и лекарственных средствах.
Алкалоидами называют группу азотсодержащих органических соединений, преимущественно растительного происхождения, проявляющих основные свойства и высокую биологическую активность. К настоящему времени выделено более 5000 алкалоидов.

|
645 |
метаболизма пуриновых |
наркотические анальгетики, |
оснований |
жаропонижающие и |
|
противовоспалительные средства |
Всостав ДНК входят аденин, гуанин, цитозин и тимин, в состав РНК
—аденин, гуанин, цитозин, урацил.
Всостав нуклеиновых кислот могут входить гипоксантин, метильные производные урацила и гуанина, гидрированные производные урацила и др.
646
РАЗДЕЛ 4 АНАЛИТИЧЕСКАЯ ХИМИЯ
4.1ВВЕДЕНИЕ.
МЕТРОЛОГИЧЕСКИЕ ОСНОВЫ ХИМИЧЕСКОГО АНАЛИЗА
ПЛАН
4.1.1 Предмет аналитической химии.
4.1.2 Краткая история развития аналитической химии.
4.1.3 Методы аналитической химии.
4.1.4 Метрологические основы аналитической химии.
4.1.1. Предмет аналитической химии
Аналитическая химия – это наука об определении химического состава веществ и отчасти их химического строения. Однако это определение не является исчерпывающим. Предметом аналитической химии являются также разработка методов анализа и их практическое выполнение, изучение теоретических основ аналитических методов. Поэтому в более широком понимании аналитическая химия – это наука о способах идентификации химических соединений, о принципах и методах определения химического состава веществ и их химической структуры.
Изучить состав вещества – это значит определить:
1)какие химические элементы образуют данное вещество;
2)в каких количественных соотношениях элементы входят в состав вещества;
3)какие функциональные группы входят в состав вещества;
4)каков изотопный состав вещества.
Как любая наука, химия преследует свои цели и задачи:
1.Разработка теоретических основ химического анализа.
2.Разработка методов анализа.
3.Анализ различных объектов.
Таким образом, аналитическая химия является одновременно и теоретической и прикладной наукой, так как она создает новые законы и методы анализа, изучает состав и строение многих веществ. С другой стороны, развитие аналитической химии диктуется потребностями практики.
С момента зарождения аналитической химии и по настоящее время стимулами ее развития являются:
1.Человеческая любознательность. «Ученый изучает природу не потому, что это полезно; он изучает ее потому, что это доставляет ему удовольствие…» (А. Пуанкаре).
2.Практические потребности общества. «Если у общества появляется техническая потребность, то она продвигает науку вперед больше, чем десяток университетов» (Ф. Энгельс).
647
4.1.2 Краткая история развития аналитической химии
Аналитическая химия прошла длительный путь развития, начало которого относится к III–IV в. до н.э., когда люди впервые научились выделять металлы из руд, получать сплавы и определять в них содержание золота и серебра.
Вот некоторые даты первых открытий в области анализа веществ:
310 г. до н.э. – Теофраст (Греция) описал способы проверки чистоты золота (на пробирном камне и с помощью «испытания огнем»);
242 г. до н.э. – Архимед (Сиракузы) проанализировал сплав золота и серебра, измерив его плотность;
20 г. н.э. – Витрувий (Древний Рим) изложил методику гравиметрического определения минерализации природных вод;70 г. н.э. – Плиний (Рим) описал пробирную плавку и качественные реакции в растворах (проверка качества квасцов).
Приемы обнаружения и определения металлов (обычно драгоценных) в рудах и выявление примесей в изделиях из этих металлов (пробирное искусство) начали формироваться в Античности, полностью сложились в Средневековье, а расцвет их пришелся на XVI век.
Пробирным искусством занимались ремесленники (рудознатцы, кузнецы, ювелиры, специалисты – «пробирщики»), а также торговцы. Знания передавались в устной форме, записываться стали только в XIV–XVII вв.
К началу XIX в. пробирное искусство было вытеснено химическими методами анализа, но отдельные его приемы применяются аналитиками до сих пор.
В 1343 г. впервые по указу короля Франции Филиппа VI Валуа была стандартизована и официально утверждена на национальном уровне методика химического анализа. В указе содержался способ определения золота в монетных сплавах с применением купелирования. Были регламентированы масса пробы, количество реагентов, детально описаны все операции и требования к весам. За любое отступление от этой методики полагалось длительное тюремное заключение.
Алхимия и анализ. Алхимики не изучали реальный состав веществ, но они создали предпосылки для формирования химических методов анализа. В этот период возникла идея о возможности направленного превращения веществ путем проведения химических реакций. Были разработаны техника и приемы химического эксперимента, накапливались эмпирические (опытные) знания о свойствах веществ и их растворов. Алхимики начали разработку химической терминологии.
Начиная с конца XVII в., развитие химического анализа перестало быть эмпирическим и случайным. Химический анализ стал объектом деятельности ученых.
Новый период развития аналитической химии начинается с середины VIII века, ознаменованной открытием М.В. Ломоносовым закона сохранения
648
массы веществ, Аррениусом – теории электролитической диссоциации и Д.И. Менделеевым – периодического закона.
Ученые, внесшие наибольший вклад в развитие аналитической химии: Роберт Бойль (1627–1691) считается родоначальником аналитической
химии как науки. Выдвинул базовые понятия «химический анализ» и «химический элемент». Научно обосновал метод гравиметрии. Развивал технику анализа. Изобрел ряд измерительных приборов. Изучал и систематически применял качественные реакции. Открыл кислотноосновные индикаторы. Указал на зависимость интенсивности окраски растворов от концентрации растворенного вещества. Систематически исследовал химический состав минеральных вод. Ввел понятие «химический анализ».
Торберн Улаф Бергман (1735–1784). Профессор Упсальского университета (Швеция). Крупнейший химик-аналитик XVIII века.
Усовершенствовал пробирный анализ с паяльной трубкой и создал газовый анализ. Определил содержание С и Р в чугунах и сталях, что легло в основу теории металлургии. Осознал различие между качественным и количественным анализом, ввел понятие «аналитический реагент», сформулировал требования к реагентам. Начал разработку методов качественного анализа с применением групповых реагентов (H2S). Сформулировал общие принципы пробоотбора и про-боподготовки в количественном анализе. Создал гравиметрический метод осаждения и проанализировал ряд минералов.
Антуан Лоран Лавуазье (1743–1794). Член Парижской академии наук, директор Управления порохов и селитр. Создатель «новой химии». Автор ряда учебников. Лавуазье сформулировал закон сохранения массы веществ в химических реакциях и доказал его методом гравиметрического анализа, предложил вести расчеты по уравнениям химических реакций, установил качественный и количественный состав воздуха и вод. Он создал гравиметрический метод элементного анализа органических веществ (сожжение в кислороде), разработал способы проверки правильности результатов количественного анализа и повышения их точности.
Йенс Якоб Берцелиус (1779–1848). Шведский химик и минералог. Открыл ряд элементов, ввел современную систему записи химических уравнений. Создал электрохимическую теорию химической связи и на ее основе классифицировал вещества и элементы. Крупнейший химик-аналитик XIX века. Развил теорию весового анализа и разработал множество методик. Создал микроанализ и соответствующую технику лабораторных работ. Исследовал источники систематических погрешностей в весовом анализе и разработал способы их снижения. Весьма точно определил атомные массы всех известных в его время элементов (41 элемент). Лично проанализировал свыше 2000 соединений, установив их брутто-формулы, а также исследовал состав многих минералов.
649
Карл Ремигий Фрезениус (1818–1897). Немецкий химик, в 1848 г. он основал в Сельскохозяйственном институте Висбадена собственную частную химическую лабораторию, ставшую одной из лучших аналитических лабораторий Европы. Написал ставшие классическими руководства по качественному и количественному анализу. Разработал четкую схему анализа катионов и разделил металлы по отношению к сероводороду на шесть аналитических групп. Эта система была так целесообразна и хорошо продумана, что сохранила свое значение вплоть до XX века. К.Р. Фрезениус является создателем химической криминалистики. Будучи не только ученым, но и педагогом и популяризатором химических знаний, он основал «Журнал аналитической химии», впервые разработал методику преподавания аналитической химии в вузах. Автор многих учебников.
Вильгельм Оствальд (1853–1932). Один из основоположников физической химии. Автор 77 книг и 300 статей. Философ, общественный деятель, нобелевский лауреат. Создал теорию ионных реакций. Осознал различие между общими и равновесными концентрациями, предложил способы вычисления и измерения равновесных концентраций. Определил сотни констант равновесия реакций в растворах. Выдвинул идею буферных растворов. Создал ионную теорию кислотно-основных индикаторов и способы подбора индикаторов при титровании слабых электролитов.
Аналитическая химия в России развивалась на первом этапе как наука, позволяющая определить состав лекарств и руд. В 1581 г. по указу Ивана Грозного была создана первая аптека, которая выполняла анализ руд. Первая лаборатория, существование которой доказано архивными документами, была создана в 1720 г. при Берг-коллегии в Петербурге по распоряжению Петра I, который сам интересовался «пробирным искусством». Сохранились его собственноручные записи методик анализа руд, найден чертеж «пробирочной печи». Известно, что лаборатории были при крупных заводах, особенно металлургических, они имели аналитическую и технологическую направленность.
Основателем химической науки в нашей стране является Михаил Васильевич Ломоносов. Он основал первую в России химическую научноисследовательскую лабораторию.
12 октября 1748 г. М.В. Ломоносов сообщил в академическую канцелярию, что «лаборатория, которая прошедшего августа 3-го числа при Ботаническом саду заложена, приведена со всем внешним и внутренним строением к окончанию и подрядчик Михайло Горбунов по контракту все исполнил».
Химические операции, применяемые в анализе, М.В. Ломоносов описал в руководстве по металлургии, в соответствии с традициями своего времени, а в 1744 г. впервые применил микроскоп для изучения химических процессов. По его инициативе и проекту в 1755 г. был основан Московский университет – первый университет России, сыгравший огромную роль в развитии науки и образования в нашей стране.
650
Большой вклад в развитие аналитической химии внесли и многие другие российские и советские ученые.
Bасилий Mихайлович Севергин создал руководства по химическому анализу минералов, руд, минеральных вод, лекарственных препаратов, например, «Способ испытывать минеральные воды» (1800 г.), «Пробирное искусство» (1801 г.). Он предложил колориметрический метод анализа.
Александр Михайлович Бутлеров создал теорию строения органических соединений.
Николай Александрович Меншуткин организовал Русское химическое общество, которое в 1869 году стало издавать свой журнал, автор книги «Аналитическая химия» (1871 г.), переведенной на немецкий и английский язык и выдержавшей 16 изданий.
Лев Александрович Чугаев изучал комплексные соединения металлов с органическими реагентами. В результате этих исследований в 1905 г. был предложен диметилглиоксим как реагент для обнаружения никеля, известный во всем мире как реактив Чугаева.
Михаил Семенович Цвет – основатель адсорбционного метода хроматографического анализа, открывшего широчайшие возможности для тонкого химического исследования. Открытие Цвета получило широкое применение и признание с начала 1930-х годов при разделении и идентификации различных пигментов, витаминов, ферментов, гормонов и других органических и неорганических соединений, а также послужило основой для создания ряда новых направлений аналитической химии (газовая хроматография, жидкостная хроматография, тонкослойная хроматография).
Иван Павлович Алимарин – советский химик-аналитик, академик АН
СССР, профессор, заведующий кафедрой аналитической химии МГУ. Его основные научные исследования посвящены разработке методов количественного микро- и ультрамикрохимического анализа минералов, руд и металлов. Он развил теоретические представления и разработал практику определения следов примесей в веществах высокой чистоты. Впервые в
СССР применил метод нейтроно-активационного определения примесей в полупроводниках. Разработал теоретические основы разделения и определения редких элементов с применением органических реактивов. Опубликовал около 500 научных работ.
Юрий Александрович Золотов – заведующий кафедрой аналитической химии МГУ, действительный член РАН, директор Института общей и неорганической химии РАН, Президент Российского химического общества им. Д.И. Менделеева (1991–1995), главный редактор «Журнала аналитической химии». Выдающийся ученый в области аналитической химии. Развил теорию жидкость-жидкостной экстракции элементов. Ввел понятие о гибридных методах анализа. Совместно с сотрудниками разработал много методов анализа с использованием концентрирования. Под его руководством и при его активном участии решены практически важные задачи в области анализа высокочистых веществ, объектов окружающей
651
среды, а также специальные задачи. Автор более 800 научных публикаций и 30 патентов, более 30 книг – монографий, справочников, учебных пособий. Книги Ю.А. Зотова изданы на русском, английском, немецком, японском и румынском языках.
В настоящее время аналитическая химия как область науки перестала быть только частью химии, она превратилась в крупную самостоятельную дисциплину. Связано это в основном с мощным расширением арсенала методов анализа, среди которых химические, физические, биологические.
Аналитическая химия, аналитическая служба решают, или должны решать, множество жизненно важных задач в государстве и обществе. Это контроль производственных процессов, диагностика в медицине, мониторинг объектов окружающей среды, обеспечение нужд военных, криминалистов, археологов.
Развивается новая общая теория, включающая, например, метрологию анализа. Резко возросли возможности химического анализа в части чувствительности и быстроты. Многие методы позволяют одновременно определять несколько десятков компонентов.
Обычными становятся анализы без разрушения анализируемого образца, на большом расстоянии, в потоке, в отдельной микроскопической точке или на поверхности. Математизация и компьютеризация значительно расширили возможности известных методов и позволили создать принципиально новые.
4.1.3 Методы аналитической химии
Исследователь, прежде чем приступить к выполнению какой-либо аналитической задачи, строит ее абстрактную модель.
Обобщенная модель, отражающая основные этапы аналитического исследования, называется аналитическим циклом (рисунок 4.1.1) и состоит из следующих этапов:
1.Общая постановка задачи.
2.Постановка конкретной аналитической задачи.
3.Выбор принципа, метода и методики анализа.
4.Пробоотбор.
5.Пробоподготовка.
6.Проведение анализа.
7.Расчеты.
8.Обработка результатов.
9.Результат анализа
Аналитический процесс – процесс получения и переработки информации о химическом составе вещества
Принцип анализа – явление, свойство или закономерность, положенное в основу метода анализа веществ.

652
Рисунок 4.1.1 – Аналитический цикл
Метод анализа – универсальный и теоретически обоснованный способ получения информации о химическом составе вещества на основе принципа или принципов анализа (рисунок 4.1.2).
Методика анализа – подробное описание правил и операций определения состава конкретного объекта с использованием выбранных методов (т.е. методика включает всю сумму тактических шагов).
Рисунок 4.1.2 – Аналитический процесс и его стадии
Пробоотбор – процедура, заключающаяся в отборе части вещества или материала с целью формирования пробы.
Проба – небольшая часть анализируемого объекта, средний состав и свойства которой должны быть идентичны во всех отношениях среднему составу и свойствам анализируемого объекта.
653
В зависимости от способа получения различают следующие виды проб:
-точечная проба – количество вещества/материала, которое отбирается от объекта за одну операцию пробоотбора; это проба, которая отбирается непосредственно из объекта;
-генеральная (объединенная) проба – проба, получаемая объединением точечных проб, отобранных от одного материала (партии). Она может быть достаточно большой: от 1 до 50 кг, иногда даже до 5 т;
-лабораторная проба – сокращенная генеральная проба, масса которой, обычно, составляет от 25 г до 1 кг;
-аналитическая проба (проба для анализа) – сокращенная лабораторная проба, которую полностью и единовременно используют для проведения анализа.
Проба должна удовлетворять ряду требований:
1)она должна быть представительной по отношению к объекту анализа, т.е. содержание определяемого компонента в анализируемой пробе должно отражать среднее содержание этого компонента во всем объекте;
2)проба должна быть устойчивой, т.е. во время транспортировки и хранения в ней не должно протекать каких-либо химических реакций;
3)проба не должна содержать никаких загрязнений – ни из устройства пробоотбора, ни из материала контейнера, ни из консервирующего реагента;
4)проба должна быть представлена в количестве, достаточном для анализа. Количество пробы, отбираемой для анализа, определяется погрешностями пробоотбора и требуемой точностью результатов. Чем выше погрешность пробоотбора и чем выше требования к точности, тем больше должна быть проба.
Пробоподготовка – совокупность процедур, проводимых с целью подготовки пробы к анализу.
Процедура пробоподготовки обычно состоит из двух частей: предварительной и окончательной стадий.
1.Предварительная стадия, цель которой – получение пробы определенной массы и однородности. Эта стадия включает, обычно, следующие основные операции:
- высушивание: образец высушивают на воздухе или в сушильном шкафу при 105–120 оС в течение 1–2 ч; при сушке сложных объектов (растения, пищевые продукты и т.п.) используют вакуумную сушку или микроволновое излучение, что сокращает время операции до нескольких минут;
- измельчение, смешивание и т.п. Любая проба нуждается в дополнительной гомогенизации перед ее усреднением и сокращением, в противном случае ее представительность не может быть гарантирована.
2.Окончательная стадия, цель которой – переведение пробы в удобную для проведения измерений форму, т.е. такое физическое состояние, которое необходимо для выбранной методики.
654
Основные операции – растворение, вскрытие (разложение) пробы, разбавление, минерализация и др.
Растворение пробы в различных растворителях (воде, кислотах, их смесях, щелочах и органических растворителях) относят к так называемым «мокрым» способам пробоподготовки.
К альтернативному «сухому» способу прибегают, когда «мокрый» способ невозможен.
«Сухой» способ, как правило, включает:
-термическое разложение,
-сплавление и спекание с различными веществами.
Измерение аналитического сигнала. Под аналитическим сигналом
(АС) понимают сигнал, функционально связанный с химическим составом анализируемого вещества, и измеряемый в ходе выполнения методики анализа.
Измерение – совокупность операций, выполняемых для определения количественного значения величины.
Результатом измерительного процесса в ходе анализа является значение аналитического сигнала (Y). Поэтому измерение можно рассматривать как получение информации о величине (значении) аналитического сигнала.
Обработка аналитического сигнала – получение значения определяемой величины.
Для извлечения аналитической информации необходимо установить функциональное соответствие между измеряемым сигналом и определяемой величиной (концентрацией или количеством компонента в пробе).
Связь между измеряемым сигналом (Y) и определяемой величиной (X) (концентрация или логарифм концентрации определяемого компонента и др.) обычно носит линейный характер и может быть представлена уравнением:
Y = K•X, |
(4.1.1) |
где K – коэффициент, включающий величины, которым можно приписать определенный химический или физический смысл.
В аналитической химии используются методы разделения и методы определения. Основной задачей методов разделения является, главным образом, отделение мешающих компонентов или выделение нужного компонента в виде, пригодном для определения. Однако часто определение интересующего компонента производится непосредственно в пробе без предварительного разделения. В некоторых случаях методы разделения и определения настолько тесно связаны между собой, что составляют единое целое.
Классификация методов определения. В зависимости от поставленной задачи аналитическую химию делят на качественный и количественный анализ.

655
Качественный анализ заключается в обнаружении отдельных элементов (или ионов), из которых состоит анализируемое вещество.
Количественный анализ заключается в определении количественного содержания отдельных составных частей сложного вещества.
В зависимости от того, какие компоненты следует определить, различают следующие виды анализа (таблица 4.1.1).
Таблица 4.1.1 – Классификация методов анализа по объектам определения
Вид анализа |
Объект определения |
Объект анализа |
|
|
|
|
|
Изотопный |
Изотопы |
Атомная энергетика, экология, |
|
медицина, археология |
|||
|
|
||
Элементный |
Элементы |
Повсеместно |
|
|
|
|
|
Вещественный |
Форма элемента (степень |
Химическая технология, экология, |
|
окисления и др.) |
геология, металлургия |
||
|
|||
|
Состав и структура молекул в |
Медицина, химическая |
|
Молекулярный |
технология, экология, |
||
сложном материале |
|||
|
криминалистика |
||
|
|
||
Функциональный |
Совокупность молекул с |
|
|
близкими свойствами (спирты, |
Химическая технология, пищевая |
||
(структурно- |
|||
моносахариды), |
промышленность, медицина |
||
групповой) |
|||
функциональные группы |
|
||
|
|
||
Фазовый |
Отдельные фазы (графит в |
Металлургия, геология, |
|
стали) |
стройматериалы |
||
|
В зависимости от способа выполнения бывают двух видов сухие и мокрые:
Сухие – проводятся без перевода твердого вещества в раствор. Анализируемый раствор предварительно выпаривается.
Мокрые – анализируемое вещество предварительно растворяется в подходящем растворителе и затем полученный раствор подвергается анализу.
Сухие химические методы классифицируются по технике исполнения: 1. Возгонка, разложение при нагревании Пример: разложение солей аммония:
NH4Cl + t0 = NH3↑ + HCl↑
2. Порошковый метод

656
NH4Cl + Ca(OH)2 = NH3↑ + H2O + CaCl2
3. Пирохимические методы:
-Окрашивание бесцветного пламени летучими соединениями металлов;
-Образование окрашенных перлов;
-Окраска пепла.
По массе или объему анализируемого вещества мокрые методы анализа подразделяются на пять видов (таблица 4.1.2), а по природе объекта на 1) анализ неорганических веществ и 2) анализ органических веществ.
Таблица 4.1.2 – Классификация методов анализа по массе или объему вещества
Вид анализа |
Масса пробы, г |
Объем раствора, мл |
Макроанализ |
>0,1 |
10-1000 |
Полумикроанализ |
0,01-0,1 |
0,1-10 |
Микроанализ |
<0,01 |
0,01-1 |
Субмикроанализ |
0,0001-0,001 |
<0,01 |
Ультрамикроанализ |
<0,0001 |
<0,001 |
Методические приемы мокрых методов: 1. Полумикроанализ:
Пробирочный – опыты проводят в пробирках, осадок отделяют центрифугированием;
Экстракционный – определяемый компонент взаимодействует с реагентом в водной фазе, продукт реакции извлекается в другой (органический) растворитель. Опыт проводят в пробирках с притертыми пробками. Экстракция осуществляется при сильном встряхивании смеси.

657
Каталитический – опыт проводят в пробирках, для ускорения реакции используют катализаторы.
2. Микроанализ:
Капельный – реакция проводится на капельной пластинке-палетке, предметном стекле или фильтровальной бумаге. Вещества добавляются по каплям.
Люминесцентный – реакция проводится капельно на предметном стекле или фильтровальной бумаге. Влажное пятно высушивается на воздухе и облучается УФ-светом. При этом наблюдается свечение (люминесценция). Требуется контрольный опыт.
Микрокристаллоскопический – реакция проводится капельно на предметном стекле. Полученный осадок рассматривается под микроскопом.
По измеряемому свойству вещества все методы химического анализа делятся на: 1) химические; 2) физические; 3) физико-химические; 4) биологические.
Химические методы анализа основаны на химических реакциях,  сопровождающихся
сопровождающихся наглядным внешним
наглядным внешним эффектом,
эффектом, 
 – выделением газа, выпадением осадка, изменением окраски. Физические методы анализа используют для изучения физических свойств вещества при помощи приборов. К ним относятся спектральные, ядерно-физические методы анализа, рентгеноструктурный анализ. Физико-химические методы основаны на измерении физико-химических свойств вещества, изменяющихся в результате химической реакции, например, потенциометрия – изменение электродного потенциала, кондуктометрия – изменение электропроводности и др. В отдельную группу следует выделить биологические методы анализа.
– выделением газа, выпадением осадка, изменением окраски. Физические методы анализа используют для изучения физических свойств вещества при помощи приборов. К ним относятся спектральные, ядерно-физические методы анализа, рентгеноструктурный анализ. Физико-химические методы основаны на измерении физико-химических свойств вещества, изменяющихся в результате химической реакции, например, потенциометрия – изменение электродного потенциала, кондуктометрия – изменение электропроводности и др. В отдельную группу следует выделить биологические методы анализа.
4.1.4 Метрологические основы аналитической химии
При анализе исследуемого образца химик-аналитик проводит обычно несколько параллельных определений, которые характеризуются двумя факторами: воспроизводимостью полученных результатов и соответствием их истинному содержанию в образце.
Воспроизводимость зависит от случайной ошибки метода анализа. Чем больше случайная ошибка, тем больше разброс значений при повторении анализа, тем меньше точность метода анализа. Отклонения от истинного содержания образца определяются систематической ошибкой. Случайные ошибки исключить невозможно, но их можно описать при помощи методов математической статистики. Эти методы исходят из представления о том, что параллельные определения, которые проводят аналитики, повторяются бесконечное число раз и составляют генеральную совокупность. Однако на практике имеется всегда очень ограниченное число полученных результатов, так называемая выборка. Если внутри серии анализов существенна только случайная ошибка, то результаты беспорядочно рассеиваются внутри небольшой области значений, несмотря
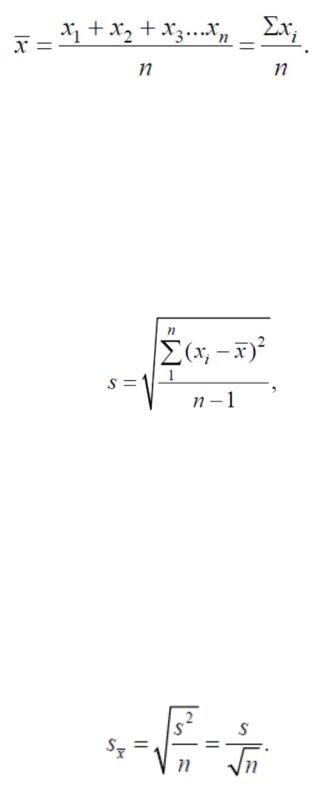
658
на совершенно постоянные условия определения. Наиболее правильное содержание пробы лежит внутри этой области колебаний.
Случайная ошибка может иметь размерность измеряемых величин; в
этом случае говорят об абсолютной ошибке определения.
Если случайная ошибка отнесена к среднему значению измеряемой величины, то в этом случае говорят об относительной ошибке определения.
При оценке результатов n анализов пользуются средним арифметическим x, которое находят по формуле
(4.1.2)
Для большого числа n определений арифметическое среднее x в основном представляет собой хорошее приближение к среднему значению в генеральной совокупности. Граница разброса отдельных измерений относительно x характеризуется квадратичной ошибкой или стандартным отклонением S. Средняя квадратичная ошибка (стандартное отклонение) выборки определяется выражением, где S мера разброса, характеризует случайную ошибку метода анализа. Средняя квадратичная ошибка S является приближением для соответствующей величины σ в генеральной совокупности.
(4.1.3)
Ее квадрат (соответственно σ2) называют дисперсией. Величина n – 1, стоящая в знаменателе равенства, называется числом степеней свободы (f). При n → ∞ x в пределе приближается к генеральной средней, а S – к стандартному отклонению совокупности σ.
Рассмотренное выше стандартное отклонение σ генеральной совокупности результатов анализа связано с вероятной ошибкой единичного наблюдения. Если из этой совокупности извлекаются серии случайных выборок объемом n анализов, то среднее значение x разных групп из n анализов будет показывать все меньшее рассеяние по мере увеличения n. При увеличении n среднее x каждой выборки в пределе приближается к генеральному среднему μ, а рассеяние стремится к нулю. Для среднего результата x стандартное отклонение определяется по формуле:
(4.1.4)

659
Если имеются результаты анализа образцов с различным содержанием, то в предположении, что средняя квадратичная ошибка не зависит от содержания, т. е. х, из частных средних квадратичных ошибок S путем усреднения можно вычислить общую среднюю квадратичную ошибку S. Если имеется m проб и если для каждой пробы проводится nj параллельных определений, то используют следующую формулу со степенями свободы f = n – m, где n – общее число анализов, n = mnj.
(4.1.5)
Когда проводят по два параллельных анализа для каждого бразца и находят значения х' и х", то для m образцов уравнение преобразуется в выражение при f = m степеней свободы:
(4.1.6)
Для данной серии анализов, проведенных тщательно и в одинаковых условиях, величина S практически не зависит от числа опытов (при большом числе опытов особенно), но часто зависит от величины содержания компонента в образце и состава его.
В последнем случае случайную ошибку иногда выражают относительной величиной, рассчитывая так называемый коэффициент вариации (V):
(4.1.7)
При многократном повторении одного анализа его результат иногда особенно сильно отклоняется в ту или иную сторону без достаточного основания. Тогда возникает вопрос, имеется ли в данном случае случайное большое отклонение Q или «грубая ошибка», которую можно в дальнейшем исключить, анализируя повторяющиеся результаты. Устанавливать грубые ошибки при небольшом числе анализов можно при помощи размаха варьирования R (разница между двумя крайними
значениями xi, xmax–xmin).
Для этого составляют отношение:

660
(4.1.8)
Где, x1 – подозрительно выделяющееся значение; х2 – соседнее с ним значение. Вычисленную величину Q сопоставляют с табличным значением Q(p, nj). Наличие грубой ошибки действительно доказано, если Q > Q(p, nj)
Результаты статистической обработки аналитических данных используются для установления числа параллельных определений, необходимых для того, чтобы средний результат имел точность, не ниже заданной. Решение этой задачи основано на упомянутом выше факте, что стандартное отклонение среднего результата S для данной серии анализов практически не зависит от числа их, тогда как sx для среднего результата зависит от n. Найдя S из небольшого числа опытов, задавшись некоторой надежностью p и учитывая требуемую точность
используем найденную выше связь перечисленных величин с помощью критерия Стьюдента t:
(4.1.9)
В последнем выражении путем последовательных подстановок подбирают такие значения n, чтобы полученный при этом коэффициент Стьюдента t (для f = n – 1) отвечал выбранной надежности p.
Если n получается слишком большим (например, более 8), это означает, что достижение заданной точности принятым методом анализа затруднительно. В таких случаях необходимо избрать другой, более точный метод
661
4.2 МЕТОДЫ ПРОБООТБОРА И ПРОБОПОДГОТОВКИ
ОСНОВНЫХ ОБЪЕКТОВ АНАЛИЗА
ПЛАН
4.2.1. Отбор проб.
4.2.1.1Отбор пробы газов.
4.2.1.2Отбор пробы жидкостей.
4.2.1.3Отбор пробы твердых веществ.
4.2.2Потери и загрязнения при отборе пробы. Хранение пробы.
4.2.3Подготовка пробы к анализу.
4.2.1. Отбор проб
Химический анализ чаще всего начинают с отбора и подготовки пробы к анализу. Следует отметить, что все стадии анализа связаны между собой. Так, тщательно измеренный аналитический сигнал не дает правильной информации о содержании определяемого компонента, если неправильно проведен отбор или подготовка пробы к анализу. В большинстве случаев именно отбор и подготовка пробы к химическому анализу лимитирует надежность и, в целом, качество получаемых результатов, а также трудоемкость и длительность аналитического цикла.
Погрешность при отборе и подготовке пробы часто определяет общую погрешность определения компонента и делает бессмысленным использование высокоточных методов и методик. В свою очередь отбор и подготовка пробы зависят не только от природы анализируемого объекта, но и от поставленной задачи и выбранного способа измерения аналитического сигнала. Приемы и порядок отбора пробы и ее подготовки настолько важны при проведении массового химического анализа, что обычно предписываются Государственным стандартом (ГОСТ).
Для проведения анализа, как правило, берут так называемую представительную (среднюю) пробу. Это небольшая часть анализируемого объекта, средний состав и свойства которой должны быть идентичны во всех отношениях среднему составу и свойствам исследуемого объекта. Различают генеральную, лабораторную и анализируемую пробы. Генеральная (называемая иногда первичной, большой или грубой) проба отбирается непосредственно из анализируемого объекта. Она достаточно большая — обычно 1-50 кг, для некоторых объектов (например, руды) составляет иногда
0,5-5 т.
Из генеральной пробы путем ее сокращения отбирают лабораторную пробу (обычно от 25 г до 1 кг). Одну часть лабораторной пробы используют для предварительных исследований, другую – сохраняют для возможных в будущем арбитражных анализов, третью – используют непосредственно для анализа (анализируемая проба). В случае необходимости пробу измельчают и усредняют. В анализируемой пробе проводят несколько определений компонента: из отдельных навесок 10-1000 мг (если анализируемый объект –
662
твердое вещество) или аликвот (если анализируемый объект – жидкость или газ).
Содержание определяемого компонента в анализируемой пробе должно отражать среднее содержание этого компонента во всем исследуемом объекте, т. е. анализируемая проба должна быть представительной. Насколько это важно, можно показать на следующих примерах. Так, при массе анализируемой пробы 1-10г оценивается среднее содержание определяемого компонента в генеральной пробе массой в несколько тонн и в конечном счете, например, запас компонента в месторождении.
Определение содержания физиологически активного компонента в анализируемой пробе из одной или нескольких таблеток дает основание для оценки эффективности всей партии лекарственного препарата. Эти примеры показывают необходимость правильного отбора пробы. Погрешность в отборе пробы часто определяет общую погрешность химического анализа и, не оценив погрешности на этой стадии, нельзя говорить о правильности определения компонента в анализируемом объекте.
Чем больше материала отобрано для пробы, тем она представительнее. Однако с очень большой пробой трудно работать, это увеличивает время анализа и расходы на него. Таким образом, отбирать пробу нужно так, чтобы она была представительной и не очень большой.
Способы отбора пробы и ее величина прежде всего определяются физическими и химическими свойствами анализируемого объекта. При отборе пробы нужно учитывать: 1) агрегатное состояние анализируемого объекта (способы отбора пробы различны для газов, жидкостей и твердых веществ); 2) неоднородность анализируемого материала и размер частиц, с которых начинается неоднородность (чем однороднее вещество, тем проще отобрать пробу); 3) требуемую точность оценки содержания компонента во всей массе анализируемого объекта в зависимости от задачи анализа и природы исследуемого объекта (так, при определении физиологически активного компонента в лекарстве требуется большая точность, чем при определении содержания компонента в руде для оценки рентабельности месторождения).
Один из факторов, который нужно учитывать при выборе способа отбора пробы, – возможность изменения состава объекта и содержания определяемого компонента во времени. Например, переменный состав воды в реке, колебания состава дымовых газов промышленного предприятия, изменение концентрации компонентов в пищевых продуктах и т. д.
Рассмотрим подробнее отбор пробы газов, жидкостей и твердых веществ.
4.2.1.1 Отбор пробы газов
Пробу газа отбирают, измеряя его объем при помощи вакуумной мерной колбы или бюретки с соответствующей запорной жидкостью, часто конденсируют газ в ловушках разного типа при низких температурах.

663
По-разному отбирают пробу газа из замкнутой емкости и из потока. В замкнутой емкости (например, цех предприятия, рабочая комната и т. д.) пробу газа отбирают в разных точках, в зависимости от задачи объемы газа смешивают или анализируют отдельно каждую пробу. При отборе пробы из потока газа обычно используют метод продольных струй и метод поперечных сечений. Метод продольных струй применяют, когда состав газа вдоль потока не меняется. В этом случае поток делят на ряд струй вдоль потока и пробы газа отбирают в струях через одну (рис. 4.2.1, а). Если состав газа вдоль потока меняется, то пробы берут на определенных расстояниях (часто через специальные отверстия в трубах) вдоль потока (рисунок 4.2.1,
б).
Рисунок 4.2.1 – Отбор пробы газа в потоке: а — метод продольных струй; б — метод поперечных сечений (стрелками показаны места отбора проб)
Так как состав анализируемых газов часто меняется во времени (например, в зависимости от графика работы предприятий, состояния атмосферы, температуры в помещениях и т. д.), то в зависимости от требуемой информации пробы усредняют или анализируют отдельно объемы газов, отобранные в разное время.
4.2.1.2 Отбор пробы жидкостей
Способы отбора гомогенных и гетерогенных жидкостей различны. Гомогенные жидкости, как и газы, отличаются высокой степенью
однородности, поэтому способы отбора пробы относительно просты. Смеси таких жидкостей, как правило, хорошо перемешиваются и также гомогенны. Пробу гомогенной жидкости отбирают при помощи пипеток, бюреток и мерных колб. Отбор пробы из общей емкости проводят после тщательного перемешивания.
Это важно, так как в поверхностном слое жидкости могут проходить различные химические реакции, меняющие состав образца. Если по какойлибо причине (например, из-за большого объема) жидкость нельзя хорошо

664
перемешать, то отбор пробы проводят на разной глубине и в разных местах емкости и, в зависимости от решаемой задачи, пробы анализируют отдельно или перемешивают.
Отбор гомогенной жидкости из потока проводят через определенные интервалы времени и в разных местах (рисунок 4.2.2, а). Для отбора проб на разной глубине используют специальные пробоотборные устройства — батометры различной конструкции. Основная часть батометра — цилиндрический сосуд вместимостью 1-3 л, закрывающийся сверху и снизу крышками. После погружения в жидкость на заданную глубину крышки цилиндра закрывают и сосуд с пробой поднимают на поверхность. Место и время отбора жидкости выбирают в зависимости от решаемой задачи.
Рисунок 4.2.2 – Отбор пробы: а — жидкости в потоке; б — гетерогенной жидкости пробоотборником с изолированными ячейками
Пробы гетерогенных жидкостей отбирают не только по объему, но и по массе. Чтобы отобрать пробу, поступают по-разному: в одних случаях жидкость гомогенизируют, в других, наоборот, добиваются полного ее расслоения. Гомогенизацию проводят, изменяя температуру, перемешивая жидкость или подвергая ее вибрации. Если гомогенизировать жидкость невозможно, то ее расслаивают и отбирают пробу каждой фазы, используя при этом специальные пробоотборники с большим числом забирающих камер (рис. 4.2.2, б). Так отбирают на анализ различные фракции продуктов и полупродуктов нефтеперерабатывающей промышленности. Обычно пробу берут после отстаивания смеси жидкостей в чанах или цистернах.
Таким образом, в зависимости от природы жидкости и решаемой задачи при анализе может меняться способ и время отбора пробы, ее размер. Заметим, что размер генеральной пробы жидкости, хотя и меняется в известных пределах, но все же обычно невелик и не превышает нескольких литров или килограммов:

665
4.2.1.3 Отбор пробы твердых веществ
При отборе генеральной, лабораторной и анализируемой пробы твердых веществ прежде всего возникает вопрос о размере пробы, который должен обеспечивать ее представительность. Оптимальная масса пробы обусловлена неоднородностью анализируемого объекта, размером частиц, с которых начинается неоднородность, и требованиями к точности анализа. Зависимость массы представительной пробы от размера (диаметра, d) неоднородных частиц проиллюстрирована ниже:
Для расчета оптимальной массы представительной пробы существует несколько приемов. Часто используют приближенную формулу РичердсаЧеччота:
Q = Kd2, |
(4.2.1) |
где Q – масса пробы, обеспечивающая ее представительность, кг; d – наибольший диаметр неоднородных частиц, мм; К – эмпирический коэффициент пропорциональности, характеризующий степень неоднородности распределения определяемого компонента в материале, он меняется в пределах 0,02-1.
Способы отбора генеральной пробы твердого вещества различны для веществ, находящихся в виде целого (слиток, стержни, прутья и т. д.) или сыпучего продукта. При пробоотборе от целого твердого объекта необходимо учитывать, что он может быть неоднороден. Например, состав массы отливки отличен от состава ее поверхности вследствие постепенного остывания металла. Так, при затвердевании чугуна его примеси оттесняются внутрь; неравномерно распределяются в слитках стали углерод, сера, фосфор. Процесс расслаивания в слитках металлов и сплавов называют ликвацией.
Учитывая возможную неоднородность целого анализируемого объекта, при отборе пробы его либо дробят, если вещества хрупкие, либо распиливают через равные промежутки, либо высверливают в разных местах слитка (рисунок 4.2.3.).

666
Рисунок 4.2.3 – Способы отбора генеральной пробы твердого вещества
Отбор пробы сыпучих продуктов тем труднее, чем неоднороднее анализируемый объект: в пробе должны быть представлены куски разного размера, полно отражающие состав образца. При отборе пробы сыпучих продуктов массу исследуемого объекта перемешивают и пробу отбирают в разных местах емкости и на разной глубине, используя при этом специальные щупы-пробоотборники. Если материал объекта транспортируется, то пробу отбирают с транспортера или желоба через равные промежутки времени, при другом способе транспортировки берут на анализ, например, каждую десятую лопату, тачку и т. д.
После отбора генеральной (или лабораторной) пробы твердого вещества осуществляют процесс гомогенизации, включающий операции измельчения (дробления) и просеивания. Пробы, содержащие крупные куски, разбивают в дробильных машинах и мельницах разного типа, пробы, содержащие меньшие куски, измельчают в шаровых мельницах и специальных ступках из закаленной инструментальной стали, состоящих из плиты – основания, закрепляющего кольца и пестика (ступки Абиха или Платгнера). Для тонкого измельчения используют фарфоровые, агатовые, яшмовые и кварцевые ступки с пестиками из такого же материала.
Так как в процессе дробления куски разного размера растираются поразному (мягкие материалы измельчаются гораздо быстрее, чем твердые), то возможны потери в виде пыли, приводящие к изменению состава пробы. Чтобы избежать этого, в процессе измельчения периодически делят крупные и мелкие частицы просеиванием, и крупные частицы растирают отдельно.
Операции измельчения и просеивания чередуют до тех пор, пока не получат достаточно растертую однородную пробу.
Следующий этап отбора пробы – усреднение, включающее операции перемешивания и сокращения пробы. Перемешивание проводят механически в емкостях (ящики, коробы и т. д.), перекатыванием из угла в угол на различных плоскостях (брезентовые полотнища, листы бумаги и т. д.), перемешиванием методом конуса и кольца (рисунок 4.2.4, а). Малые по объему пробы хорошо перемешиваются при растирании в шаровых мельницах.

667
Рисунок 4.2.4 – Перемешивание и сокращение пробы
Сокращение пробы проводят различными способами (рис. 4.2.4, б, в, г). Этот процесс, как правило, многостадийный, включающий повторное перемешивание и деление. Степень сокращения может быть определена заранее на основании расчета величины генеральной и анализируемой проб, которые получают в результате последовательного уменьшения объема анализируемого объекта.
4.2.2 Потери и загрязнения при отборе пробы. Хранение пробы
В процессе отбора и хранения пробы возможны потери определяемого компонента, внесение загрязнений, изменение химического состава, что приводит к увеличению общей погрешности анализа.
Потери в виде пыли можно в заметной степени уменьшить просеиванием пробы при измельчении. Другой возможный источник ошибок при отборе и хранении пробы – потеря летучих продуктов вследствие изменения температурного режима при хранении или разогрева при измельчении твердых образцов. Большие потери могут быть также вследствие адсорбции определяемого компонента на поверхностях емкостей для отбора и хранения пробы.
+Состав анализируемого объекта может меняться за счет проходящих в нем химических реакций (разложения компонентов, окисления их при взаимодействии с атмосферным кислородом). Например, концентрация пестицидов в растениях, почве и пищевых продуктах со временем значительно понижается вследствие их химических превращений. Погрешности, обусловленные внешними загрязнениями, особенно велики при определении примесей компонентов, их следовых количеств. Поэтому
668
при растирании образцов используют ступки из особо твердых материалов и хранят пробы в посуде из особых сортов стекла или полиэтилена. Например, пробы воды для определения кремния отбирают только в полиэтиленовые бутыли. При определении органических соединений предпочтительнее посуда из стекла.
Важными являются методы хранения и консервации пробы. В отдельных случаях для сохранения определяемого компонента его экстрагируют органическими растворителями или адсорбируют на различных твердых веществах. Пробы можно стабилизировать на несколько часов охлаждением до 0 ºС и на несколько месяцев – резким охлаждением до –20 ºС. Для консервирования определяемых компонентов добавляют разные консерванты (кислоты, образующие комплексные соединения вещества и др.). Хранят пробы в условиях, гарантирующих постоянство их состава в отношении тех компонентов, которые предполагается определять, при этом учитывают комплекс условий (температура, освещенность, материал посуды
ит. д.).
4.2.3Подготовка пробы к анализу
Подготовка пробы – важный этап проведения химических анализов. При подготовке пробы к анализу можно выделить три основные стадии:
1)высушивание;
2)разложение (чаще с переведением пробы в раствор);
3)устранение влияния мешающих компонентов;
4)перевод пробы в форму, требующуюся для метода определения. Высушивание пробы. Анализируемый образец содержит, как правило,
переменное количество воды. Это может быть химически несвязанная вода, например, адсорбированная на поверхности пробы твердого вещества, сорбированная щелями и капиллярами аморфных веществ (крахмал, белок), окклюдированная полостями минералов, руд, горных пород. Анализируемый объект может также содержать химически связанную воду. Это может быть кристаллизационная (например, в соединениях BaCl2·2H2O, CaSO4·2H2O, Na2B4O7·10H2O) или конституционная вода, выделяющаяся в результате разложения вещества при нагревании. Часть химически связанной воды может теряться в процессе отбора и хранения пробы.
Для установления состава объекта и получения воспроизводимых результатов необходимо удалить влагу из образца, высушив его до постоянной массы. Чаще всего анализируемый образец высушивают на воздухе или в сушильных шкафах при температуре +105 +120 ºС в течение 1–2 ч или в эксикаторах над влагопоглощающими веществами (прокаленный хлорид кальция, фосфорный ангидрид). Длительность и температуру высушивания образца, зависящие от его природы, устанавливают заранее методом термогравиметрии. Воду определяют гравиметрически косвенным или прямым методом. В косвенном методе о содержании воды судят по потере массы анализируемой пробы при ее высушивании или прокаливании.
669
Прямой гравиметрический метод основан на поглощении выделившейся из образца воды подходящим поглотителем. О содержании воды судят по увеличению массы предварительно взвешенного поглотителя.
Для определения воды также применяют титриметрический метод, газожидкостную хроматографию и инфракрасную спектроскопию.
Разложение образцов. Переведение пробы в раствор. Способы разложения делят на сухие и мокрые. К сухим относят термическое разложение, сплавление и спекание с различными веществами (солями, оксидами, щелочами и их смесями), к мокрым – растворение анализируемой пробы в различных растворителях.
Растворитель должен растворять пробу быстро, в достаточно мягких условиях и не мешать на последующих стадиях анализа. Лучшим растворителем является вода. Для растворения органических соединений применяют органические растворители (спирты, хлорированные углеводороды, кетоны). В отдельных случаях используют смесь воды и смешивающегося с ней органического растворителя (например, смесь воды и этанола).
При мокром способе разложения пробы часто применяют различные кислоты высокой степени очистки и их смеси при нагревании с использованием сосудов из соответствующего (инертного к кислотам) материала. Лучшим растворителем для многих металлов является соляная кислота. Для ускорения разложения кислотами иногда используют катализаторы (например, ферменты). Для обеспечения разложения веществ, не взаимодействующих с реагентами при обычной температуре и давлении, растворение проб часто проводят в автоклавах.
Выбор сухого способа разложения (сплавление, спекание и термическое разложение) определяется задачей анализа, природой разлагаемого вещества, выбранным методом определения компонентов, наличием необходимой аппаратуры.
Сплавление как метод разложения пробы сухим способом чаще используют при анализе неорганических веществ.
При сплавлении тонко измельченный образец перемешивают с 8–10- кратным избытком реагента (плавня) и нагревают (+300 +1 000 °С) до получения прозрачного сплава. Сплавление считается законченным, когда масса в тигле становится совершенно однородной, прозрачной и легкоподвижной. После охлаждения застывшую массу растворяют в воде или кислотах. При сплавлении используют щелочные, кислые и окислительные плавни.
Спекание – это взаимодействие веществ при повышенной температуре в твердой фазе, основанное на высоком химическом сродстве компонентов пробы к введенным реагентам, на диффузии и реакциях обмена. В отдельных случаях спекание позволяет провести разложение пробы быстрее и проще, способствует уменьшению количества загрязнений, поскольку при этом часто используют меньший (двухили четырехкратный) избыток реагентов и
670
менее высокие температуры. Спекание проводят обычно со смесью карбонатов щелочных металлов и оксидов магния, кальция или цинка. Рекомендуется использовать спекание при разложении проб силикатов, сульфидов, оксидов металлов.
Сухое озоление (термическое разложение, сожжение) наиболее распространено при вскрытии проб органического происхождения в токсикологическом анализе следовых содержаний примесей металлов. Сухое сожжение органических веществ проводят под действием кислорода воздуха или кислорода из баллона. Большинство пищевых продуктов сгорает при температуре +550 +600 °С.
Преимуществом сухого озоления является простота аппаратуры (термопечи и тигли), минимум внимания оператора, отсутствие загрязнений от реактивов; недостатком – возможность потерь легколетучих элементов (Hg, As, Se, Те), взаимодействие с материалом тигля и длительность процесса. Широкое распространение получило сухое сожжение с озоляющими добавками (окислители, разбавители, плавни, вещества, препятствующие улетучиванию элементов).
Сухой способ используют тогда, когда мокрый способ не дает удовлетворительных результатов, поскольку возрастает вероятность и величина погрешностей, особенно при сплавлении.
Пиролиз – процесс термического разложения в отсутствие веществ, реагирующих с разлагаемым соединением. При пиролизе органических веществ характеристические фрагменты органических соединений появляются главным образом в интервале +300 +700 °С. Неорганические вещества разлагаются, как правило, при температурах +1 000 +1 500 °С.
Пиролиз желательно проводить в атмосфере инертного газа (азот, гелий) или в вакууме при большой скорости нагрева. Его проводят различными способами: прокаливают пробу в тигле или небольшой лодочке в печи, наносят образец на металлическую проволоку или спираль и нагревают их до нужной температуры, помещают вещество в вакуумированную или заполненную инертным газом стеклянную или кварцевую трубку и также нагревают ее до необходимой температуры. Кроме того, применяют облучение лазером, потоком электронов высокой энергии, нагревание смеси пробы с ферромагнитным материалом (например, с порошком железа) в высокочастотном электрическом поле и т. д.
Пиролиз чаще используют при анализе органических веществ, особенно полимеров. Газообразные продукты пиролиза определяют различными аналитическими методами (газовая хроматография, ультрафиолетовая (УФ-) и инфракрасная (ИК-) спектроскопия, массспектрометрия).
Высокоэффективным способом окислительной минерализации является разложение образцов с помощью низкотемпературной кислородной плазмы, предполагающее пропускание газообразного кислорода под давлением 133–665 Па через высокочастотное электрическое
671
поле. Этот способ успешно используют для определения Zn, Cd, Pb и Cu методом дифференциальной инверсионной вольтамперометрии наряду с методом мокрого озоления в смеси хлорной и азотной кислот. Достоинствами метода являются отсутствие опасности загрязнения пробы материалом сосуда или реагентами, а также селективность (отделение органической части от неорганической), что важно при анализе почв, медико-биологических образцов, объектов животного и растительного происхождения.
При микроволновом разложении пробы источником тепла для мокрой минерализации веществ является энергия микроволнового (МВ) излучения (300–30 000 МГц), приводящая к быстрому разогреву всего объема образца, поглощающего МВ-энергию. В результате вместо 1–2 ч для полного разложения проб кислотой требуется 10–15 мин, а температура кипения достигается в течение 2 мин.
Современные способы измерения температуры и давления непосредственно в МВ-печи позволили определить температуры разложения основных компонентов пищевых продуктов азотной кислотой под давлением (углеводы – 140 °С, белки – 150 °С, жиры – 160°С). Достаточно 10 мин для полного разложения азотной кислотой всех компонентов пищевых продуктов. Использование МВ-печей позволяет автоматизировать процесс подготовки пробы и значительно ускорить ход анализа. При разложении различных проб в микроволновом поле в большинстве случаев используют
смесь (НNО3 +H2O2).
Использование ультразвука в подготовке пробы. При ультразвуковой (УЗ) обработке пробы происходит дробление частиц, увеличение поверхности перемешивания, образование эмульсий с большой поверхностью контакта. УЗ-обработка в подготовке проб пищевых продуктов и объектов окружающей среды применяется для перемешивания и измельчения материалов.
Фотохимическая подготовка пробы широко используется при определении органических веществ, углерода, азота и фосфора, присутствующих в воде. За последние годы увеличилось применение ультрафиолета в подготовке проб биологических объектов и пищевых продуктов. Особое место занимает УФ-минерализация органических веществ в катодной адсорбционной вольтамперометрии.
Электрохимический метод подготовки пробы основан на том, что в присутствии обычно хлорид-ионов ведется прямое анодное окисление органических веществ либо косвенное их окисление через реакции с частицами генерированных окислителей. Преимуществом этого метода является минимальное загрязнение проб из-за отсутствия окисляющих реактивов и возможность совмещения подготовки пробы с определением тяжелых металлов. Данный метод эффективен при обработке проб, содержащих органические вещества в малых количествах, например, в природных водах.
672
Экстракция. Для извлечения из проб пищевых продуктов органических веществ используется экстракция – процесс распределения вещества между двумя или более несмешивающимися фазами. С целью усиления экстракции в одну из фаз экстракционной системы вносят экстрагент. При анализе пищевых продуктов в качестве экстрагентов используют воду, спирты, бензол, ацетон, дихлорметан и др. Выбор экстрагента зависит от природы пищевых продуктов. Экстракционный способ имеет недостаток – необходимость отгонки значительных объемов растворителя, что может привести к потерям веществ, особенно летучих или образующих с растворителем азеотропные смеси.
Жидко-жидкостная экстракция (ЖЖЭ) – классический способ извлечения пестицидов из водных образцов при использовании дихлорметана. В настоящее время появилась микроЖЖЭ – экстракция из большого объема воды (400 мл) очень малым объемом растворителя (500 мкл), которая применяется для подготовки пробы для анализа методом газовой хроматографии без стадии испарения, что важно для определения высоколетучих соединений. В сравнении с твердофазной экстракцией данный метод подготовки пробы является более быстрым и дешевым.
Твердофазная экстракция применяется при анализе природных вод, пестицидов и продуктов их распада. Ее преимущества – экономия времени и растворителей, исключение опасности образования эмульсий, возможность выделения следовых количеств аналита и автоматизации.
Сверхкритическая жидкостная экстракция является относительно новым методом, применяемым для извлечения веществ с помощью специальных экстрагентов – «сверхкритических» жидкостей (жидкие СО2, NH3, пропан, бутан и др.). Сверхкритическая жидкостная экстракция используется для анализа пестицидов в почвах, тканях растений и животных.
673
4.3 МЕТОДЫ ОБНАРУЖЕНИЯ И ИДЕНТИФИКАЦИИ
ПЛАН
4.3.1Аналитическая реакция и её характеристики. Дробный и систематический анализ.
4.3.2Классификация катионов и анионов.
4.3.3Химические и инструментальные методы идентификации катионов и анионов.
4.3.1 Аналитическая реакция и её характеристики
Химические методы анализа основаны на проведении аналитической реакции между определяемым веществом Х и каким-либо реагентом R:
X+R↔Y.
Внешние эффекты, по которым можно судить о протекании той или иной реакции, называются аналитическим сигналом. Происходящие изменения называются аналитической реакцией, а вещества, вызвавшие эти изменения, – химическими реагентами.
Химические методы можно применять в растворах («мокрый путь» анализа), или с твердыми веществами без использования растворителя («сухой путь» анализа). «Сухим путем» осуществляют пирохимический анализ: твердое вещество нагревают, летучие соли окрашивают пламя в соответствующий цвет или сплавляют твердые вещества и получают «перлы» разной окраски.
Качественный анализ заключается в обнаружении отдельных элементов или ионов, входящих в состав анализируемого вещества, с помощью аналитических реакций.
Аналитическими являются только те реакции, которые сопровождаются внешним эффектом. Это может быть:
-выделение газа;
-изменение окраски раствора;
-выпадение осадка;
-растворение осадка;
-образование кристаллов характерной формы.
Аналитические реакции должны:
-Протекать с большой скоростью.
-Равновесие должно устанавливаться за несколько секунд, или, в крайнем случае, за несколько минут.
-При установившемся равновесии степень превращения реагирующих веществ должна быть высокой, желательно 100%.
-Реакция должна соответствовать определенному стехиометрическому коэффициенту.
674
Аналитические реакции должны соответствовать определенным требованиям:
1. Среда раствора. Например, AgCl растворяется в NH3 –
AgCl + 2NH4ОН ↔ [Ag(NH3)2]Cl + 2H2О,
поэтому он может быть получен преимущественно в кислой среде.
2.Температура. Например, NH4+ обнаруживают действием щелочи на исследуемый раствор при нагревании.
3.Концентрация анализируемого вещества.
Важными характеристиками аналитической реакции являются: предел обнаружения, чувствительность, селективность.
По значению концентрации определяемого вещества различают реакции высокочувствительные и малочувствительные.
Для оценки чувствительности используются две количественные характеристики:
1. Предел обнаружения (открываемый минимум) – это минимальная концентрация или минимальное количество вещества, которое может быть обнаружено данным методом с допустимой погрешностью. При анализе растворов обязательно указывают предел обнаружения в определённом объёме. Для аналитических реакций, проводимых в обычных пробирках, предел обнаружения указывают для объёма 1 мл. Для микрокристаллоскопических реакций объём равен 0,05 мл.
Предел обнаружения не является постоянной величиной. Он зависит от условий проведения реакции, от кислотности среды, от концентрации реактива, от присутствия примесей, от температуры и т.п. Хорошим считается предел обнаружения 10-7 г (0,1 мг) в 1 мл раствора.
Физические методы позволяют снизить предел обнаружения в твёрдых образцах до 10-15 г. Метод лазерной спектроскопии позволяет селективно обнаружить десятки отдельных атомов, а метод электронной микроскопии в некоторых случаях позволяет обнаружить отдельные атомы.
Для обеспечения низкого предела обнаружения используют следующие приёмы:
1)микрокристаллоскопический анализ;
2)капельный анализ;
3)флотация;
4)экстракция;
5)метод «умножающихся реакций» – ряд последовательных реакций, в результате которых получается новое вещество в количестве, во много раз превышающем первоначальное количество обнаруживаемого вещества:
например: 1) Cl- + AgIO3 = AgCl + IO3-;
2)IO3- + 5I- +6H+ = 3I2 + H2O.
6)каталитические реакции;
675
7)люминесцентные реакции;
8)реакции на носителях.
2. Минимальная определяемая концентрация (предельное разбавление) показывает, при какой концентрации раствора (разбавлении) реакция еще дает положительный результат.
Избирательность, или селективность – следующая важная характеристика аналитической реакции. От этой характеристика зависит, много ли других веществ будут мешать обнаружению и определению искомого вещества.
В зависимости от этой характеристики различают специфические и избирательные реакции:
1.Селективные (избирательные) – реакции, которые дают сходные эффекты лишь с ограниченным числом ионов.
Например, Cu2+, Hg2+, Pb2+ и другие образуют с I– осадки. Чем меньше таких ионов, тем выше избирательность.
2.Специфические – реакции, которые позволяют обнаружить ион в присутствии других ионов.
Примером реакции с участием специфического реагента является
выделение газообразного NH3 при действии сильных оснований (KOH или NaOH) на вещество, содержащее ион NH4+. Ни один ка-тион не помешает обнаружению иона NH4+, потому что только он ре-агирует со щелочами с выделением NH3.
Реакции в растворах бывают преимущественно между ионами, поэтому аналитические реакции позволяют обнаружить не вещество, а ионы.
К реагентам (R) предъявляют специальные требования:
1.Они должны обладать высокой степенью чистоты (содержать минимальное количество примесей). В анализе в основном используют R:
- «Химически чистый» – «х.ч.». Содержание основного компонента более 99 %.
- «Чистый для анализа» – «ч.д.а.». Содержание основного компонента до 99,9 %.
- «Особо чистый» – «о.с.ч.». Такая квалификация установлена для веществ высокой чистоты. «О.с.ч.» содержат примеси в таком незначительном количестве (менее 0,1%), что они не влияют на основные специфические свойства веществ.
2.Реагенты должны быть максимально доступны, недороги, нелетучи, безопасны.
3.Должны обладать необходимой реакционной способностью. Реагенты бывают:
1) Общими. К общим реагентам относят те, которые взаимодействуют
со многими ионами. Например, реакции взаимодействия со щелочами, растворами аммиака, кислотами, растворимыми фосфатами.
2. Групповыми. Групповые реагенты служат для отделения одной группы ионов от другой. Групповой реагент должен удовлетворять
676
следующим требованиям:
-все ионы, входящие в состав данной группы, под действием группового реагента должны полностью переходить в осадок;
-осадок ионов одной группы не должен даже частично захватывать ионы других групп;
-в составе группового реагента не должно быть ионов, присутствие которых собираются проверять в ходе дальнейшего анализа;
-полученный осадок должен легко растворяться при изменении рН;
-избыток добавленного реагента не должен мешать дальнейшему анализу, или должен легко удаляться из раствора.
Групповыми реагентами являются карбонат аммония, сульфид аммония, сероводород и другие.
3.Селективными. Селективными реагентами являются такие реактивы, которые взаимодействуют с небольшим числом ионов. Селективность реагента можно повысить одним из приемов:
- выбрав оптимальное значение рН; - переведя продукт реакции в другую фазу;
- маскируя мешающий компонент, переведя его в форму, не мешающую основной аналитической реакции.
4.Специфическими. Специфическими реагентами являются такие реагенты, которые взаимодействуют с одним ионом. Специфических реактивов очень немного. Примером может служить щелочь, применяемая для обнаружения ионов аммония, а также крахмал, позволяющий открывать иод.
NH4+ + OH- = NH3↑ +H2O.
Есть два вида качественного анализа:
1.Дробный анализ. Не предполагает предварительного разделения компонентов пробы. В дробном методе используют специфические реагенты, которые позволяют обнаружить любой ион в присутствии других ионов. Ионы обнаруживают в отдельных порциях исследуемого раствора, добавляя соответствующие реагенты.
Для дробного метода характерны следующие особенности:
1) определяемый ион обнаруживают в любой последовательности в отдельных пробах;
2) исключаются процессы выпаривания и прокаливания и промывание осадка, так как обычно анализируют фильтрат.
Недостатки:
1) мало специфических реагентов; 2) чем меньше селективность реакций, тем больше ошибка.
2.Систематический анализ. При систематическом анализе используют групповые реагенты. Из пробы сложного состава последовательно выделяют более простые по составу группы – фракции.
В зависимости от состава смеси применяют разные методы фракцио-
677
нирования – осаждение, экстракцию, сорбцию, возгонку, хроматографическое разделение. Каждый ион обнаруживают после того, как удалены другие ионы, мешающие своим присутствием.
Недостатки:
1)громоздкость и длительность выполнения анализа;
2)потери обнаруживаемых ионов, если они находятся в малых количествах.
Реакции обнаружения отдельных ионов чередуются с реакциями отделения их друг от друга. Таким образом, при систематическом методе анализа наряду с реакциями открытия отдельных ионов приходится прибегать также к реакциям отделения их друг от друга. Для разделения пользуются различиями в растворимости аналогичных соединений разделяемых ионов или различиями в летучести соответствующих соединений (выпаривают растворы).
При использовании систематического метода анализа катионы и анионы делят на группы (смотрите ниже).
4.3.2 Классификация катионов и анионов
В ходе систематического анализа катионы и анионы предварительно делят на аналитические группы. Для этого используют групповые реагенты (смотрите выше).
Аналитическая группа – группа ионов, которые с каким-либо одним реактивом при определённых условиях могут давать сходные аналитические
реакции. |
|
|
|
|
|
Основные |
классификации |
катионов |
– |
кислотно-основная, |
|
сероводородная, |
аммиачно-фосфатная |
и |
др. |
Кислотно-основная |
|
классификация основана на различной растворимости гидроксидов, хлоридов, сульфатов.
Групповыми реактивами этого метода являются растворы кислот и оснований.
По кислотно-основной классификации катионы делят на шесть аналитических групп (таблица 4.3.1).
Таблица 4.3.1 – Кислотно-основная классификация катионов
№ группы |
Катионы |
Групповой реагент |
|
|
|
1 |
Li+, Na+, K+, NH4+ |
Нет |
2 |
Ag+, Hg22+, Pb2+ |
HCl |
3 |
Ca2+, Sr2+, Ba2+ |
H2SO4 |
4 |
Zn2+, Al3+, Sn2+, Sn4+, As3+, As5+, Cr3+ |
NaOH в присутствии H2O2 |
|
|
|
5 |
Mg2+, Sb3+, Sb5+, Bi3+, Mn2+, Fe2+, Fe3+ |
NaOH |
6 |
Cu2+, Cd2+, Hg2+, Co2+, Ni2+ |
25 %-ный NH4ОH |

678
На рисунке 4.3.1 представлена схема разделения катионов по кислотноосновной классификации.
Рисунок 4.3.1 – Схема разделения катионов по кислотно-основной классификации
В качестве групповых реагентов в сероводородном систематическом методе анализа применяют карбонат аммония, сульфид аммония, сероводород и хлороводородную кислоту. Все катионы в рамках сульфидной классификации делят на 5 групп (таблица 4.3.2).

680
Аммиачно-фосфатная классификация катионов – это бессероводородная классификация, созданная в XX в. Она основана на растворимости фосфатов в воде, NH4OH, уксусной кислоте. Включает 5 аналитических групп катионов (таблица 4.3.3).
Таблица 4.3.3 – Аммиачно-фосфатная классификация катионов
№ группы |
Катионы |
Групповой реагент |
|
|
|
|
|
1 |
Na+, K+, NH4+ |
Нет |
|
2 |
Li+, Mg2+, Ca2+, Sr2+, Ba2+, Mn2+, Fe2+, Al3+, Bi3+, |
(NH4)2HPO4 в 25%-ном |
|
Cr3+, Fe3+ |
NH4OH |
||
|
|||
3 |
Cu2+, Zn2+, Cd2+, Hg2+, Co2+, Ni2+ |
(NH4)2HPO4 или Na2HPO4 |
|
4 |
Sn2+, Sn4+, Sb3+, Sb5+, As3+, As5+ |
HNO3 |
|
5 |
Ag+, Hg22+, Pb2+ |
HCl |
Аналитические классификации анионов разработаны менее подробно, чем классификации катионов. Общепризнанной классификации анионов до настоящего времени не существует. Чаще всего принимают во внимание растворимость солей бария и серебра тех или иных анионов (таблица 4.3.4) и их окислительно-восстановительные свойства в водных растворах (таблица
4.3.5).
Таблица 4.3.4 – Классификация анионов, основанная на растворимости солей
Ba2+ и Ag+
№ группы |
Анионы |
Групповой реагент |
|
|
|
|
|
1 |
SO42–, SO32–, S2O32–, C2O42–, CO32–, |
BaCl2 в нейтральной или слабо |
|
B4O72– (BO2–), PO43–, AsO43–, AsO33–, F– |
щелочной среде |
||
|
|||
2 |
Cl–, Br–, I–, BrO3–, CN–, NCS–, S2– |
AgNO3 в присутствии 2 н.HNO3 |
|
3 |
NO2–, NO3–, CH3COO– |
Нет |
В любом случае удаётся логически разделить на группы только часть известных анионов, так что всякая классификация ограничена и не охватывает все анионы, представляющие аналитический интерес. Наиболее часто используются две классификации.
681
Таблица 4.3.5 – Классификация анионов, основанная на их окислительновосстановительных свойствах
№ группы |
Анионы |
Групповой реагент |
|
|
|
|
|
1 |
BrO3–, AsO43–, NO3–, NO2– |
KI в сернокислой |
|
Анионы-окислители |
среде |
||
|
|||
2 |
S2–, SO32–, S2O32–, AsO33– |
I2 |
|
Анионы- |
S2–, SO32–, S2O32–, AsO33–, NO2–, C2O42– |
KMnO4 в |
|
восстановители |
, Cl–, Br–, I–, CN–, NCS– |
сернокислой среде |
|
3 |
SO42–, CO32–, PO43–, CH3COO–, B4O72– |
|
|
Индифферентные |
Нет |
||
(BO2–) |
|||
анионы |
|
||
|
|
Систематический анализ смеси анионов с использованием любой классификации никогда не проводится. Групповой реагент применяют для доказательства присутствия или отсутствия в смеси анионов той или иной аналитической группы. После этого намечают и реализуют наиболее целесообразную схему анализа данного конкретного объекта.
4.3.3 Химические и инструментальные методы идентификации катионов и анионов
1. Ионы NH4+
NH4+ + OH– → NH3↑ + H2O посинение лакмусовой бумажки Реактив Неслера
NH4+ + 2[HgI4]2– + 4OH– → [NH2Hg2O]I + 7I-+2Н2О
2. Ионы Na+
Микрокристаллоскопическая реакция с уранилацетатом натрия
CH3COONa + (CH3COO)2UO2 = CH3 COONa·(CH3 COO)2UO2 – осадок зеле-новато-желтого цвета и характерные тетраэдры и октаэдры.
Реакцию можно проводить с цинкуранилацетатом ZnAc2·3UO2Ac2. Идентификации натрия не мешают NH4+, Mg2+, K+, Ca2+, Ba2+, Sr2+.
Метод пламенной фотометрии Аналитический сигнал – свет, испускаемый атомами при переходах
их внешних электронов.
Длина волны λ – качественная характеристика. Качественный анализ основан на зависимости ∆E = hν.
Интенсивность излучения I – количественная характеристика сигнала, зависящая от концентрации определяемого элемента С:
I = k·C

682
Схема пламенного фотометра представлена на рисунке 4.3.3.
Рисунок 4.3.3 – Схема пламенного фотометра
Окраска пламени:
Na – желтый цвет K – фиолетовый
Sr – карминово-красный Ba – желто-зеленый
Ca – кирпично-красный Li – красный
Rb, Cs – фиолетовый
B, Cu, Bi – зеленый Pb, As, Sb – голубой
Атомно-эмиссионный метод используют для качественной идентификации элементов на стилоскопе, при этом источником света, в котором происходит атомизация, возбуждение атомов и испускание света, является электрический разряд. При наличии в образце натрия в эмиссионном спектре наблюдается яркий желтый дублет (две близко расположенные линии) при 589 нм.
3. Ионы K+
2K+ + Na+ + [Co(NO2)6]3- → K2Na[Co(NO2)6]↓ – желтый осадок Мешают ионы аммония, устранить их мешающее действие можно
отгонкой солей аммония t0
NH4Cl→ NH3↑ + HCl
4. Ионы Ca2+
Ca2+ + SO42– + 2H2O → CaSO4·2H2O – гипс,
микрокристаллоскопическая реакция (звездочки).
5. Ионы Ag+
Ag+ + Cl– → AgCl↓ – белый осадок
AgCl↓ + HCl → HAgCl2, избыток осадителя вреден
683
AgCl↓ + 2NH3 → [Ag(NH3)2]Cl
[Ag(NH3)2]Cl + KI → AgI↓ + 2NH3 + KCl – желтый осадок
6. Ионы Fe2+
3Fe2+ + 2[Fe(CN)6]3– → Fe3[Fe(CN)6]2↓ – турнбулева синь,
осадок красная кровяная соль
7. Ионы Fe3+
4Fe3+ + 3[Fe(CN)6]4- → Fe4[Fe(CN)6]3↓ берлинская лазурь,
осадок желтая кровяная соль
В действительности выпадающий синий осадок имеет один и тот же состав с плавающим зарядом:
KFe[Fe(CN)6]
III |
II |
II |
III |
Fe3+ + 4SCN– → [Fe(SCN)4]–, раствор окрашен в кроваво-красный цвет; реакция используется при фотометрическом определении железа(III).
8.Ионы Cu2+
Cu2+ + 4NH3 → [Cu(NH3)4]2+ сине-фиолетовое окрашивание раствора; сходный по цвету аммиакат дает Ni2+
Cu2+ + Fe → Cu↓ + Fe2+ – красный осадок металлической меди
9. Ионы Ni2+
[Ni(NH3)6]2+ + 2H2Dm → Ni(HDm)2↓ + 2NH4+ + 4NH3
10. Ионы Co2+
Co2+ + 4SCN– этанол→ [Co(SCN) 4]2– – синее окрашивание раствора; необходимо маскирование железа(3+)
11.Ионы Cr(3+)
2[Cr(OH)4]– + 2OH– + 3H2O2 → 2CrO42– + 8H2O – желтое окрашивание
12. Ионы Mn2+ идентифицируют окислением их с помощью PbO2, (NH4)2S2O8, NaBiO3. Реакции мешают хлорид-ионы:
2MnO4– + 16H+ + 10Cl– → 2Mn2+ + 8H2O + 5Cl2↑
684
Окисление персульфатом аммония проводят в присутствии катализатора – солей серебра:
2Ag+ + S2O82– → 2Ag2+ + 2SO42–
Далее ионы Ag2+ окисляют ионыMn2+, а сами восстанавливаются:
Mn2+ + 5Ag2+ + 4H2O → MnO4– + 5Ag+ + 8H+
Суммарно:
2Mn2+ + 5S2O82– + 8H2O → 2MnO4– + 10SO42– + 16H+
Реакция окисления висмутатом натрия:
2Mn2+ + 5BiO3– + 14H+ → 2MnO4– + 5 Bi3+ + 5Na+ + 7H2O
Для проведения берут одну каплю(!) исследуемого раствора, чтобы не протекала реакция диспропорционирования Mn2+ → MnO2↓ ← MnO4–.
13. Ионы Bi3+ обнаруживают по протеканию реакции гидролиза:
Bi3+ + Cl– + H2O → BiOCl↓ + 2H+ – белый осадок Реакция с тиомочевиной:
Bi3+ + 3(NH2)2CS → [Bi(SC(NH2)2)3]3+ – желтый комплекс
14.Ионы CO32–, HCO3–
CO32– + 2H+ → H2CO3 (H2O + CO2↑) – выделение углекислого газа
Ca(OH)2 + CO2 → CaCO3↓ + H2O – помутнение известковой воды
15. Ионы H2PO4–, HPO42-, PO43- реакция с модибденовой жидкостью
PO43– + 3NH4+ + 12MoO42– + 24H+ → (NH4)3[PMo12O40]↓ + 12 H 2O
желтый осадок фосфоромолибдата аммония
Еще один метод идентификации c магнезиальной смесью:
Mg2+ + HPO42– + NH4OH → MgNH4PO4↓ + H2O – осадок белого цвета
16. Ионы SO42–
SO42– + Ba2+ → BaSO4↓ – белый кристаллический осадок, не растворим в HCl, HNO3
17. Ионы Cl–

685
Cl– + Ag+ → AgCl↓ – белый творожистый осадок, растворимый в аммиаке
AgCl↓ + 2NH3 → [Ag(NH3)2]+ + Cl–
Подкисление [Ag(NH3)2]+ + Сl– + 2H+ → AgCl↓ + 2NH4+
18. Хлор активный
Cl2 + 2KI → 2KCl + I2 – синее окрашивание с крахмалом и фиолетовое в вазелиновом масле
19. Ионы NO3– обнаруживают с дифениламином по появлению синего окрашивания:
Идентификацию и количественное определение можно также проводить с помощью ион-селективного электрода:
Е= Е° – 0,059 lg a(NO3–)
20. Ионы СН3СОО–
СН3СООNa + H2SO4 → 2CH3COOH + Na2SO4 – запах уксуса
При добавлении этилового спирта образуется летучий эфир с характерным запахом (этилацетат).
Для определения ацетат-ионов можно использовать реакцию с хлоридом железа(III):
FeCl3 + 3CH3COONa → Fe(CH3COO)3 + NaCl
3Fe(CH 3COO)3 + 2H2O → [Fe3 (CH3COO)6(OH)2]CH3COO + 2CH3COOH, выпадает красно-бурый осадок комплексного соединения при
нагревании раствора.

686
4.4 РЕАКЦИИ И ПРОЦЕССЫ, ИСПОЛЬЗУЕМЫЕ В АНАЛИТИЧЕСКОЙ ХИМИИ
ПЛАН
4.1Кислотно-основное равновесие.
4.2Равновесие в растворах с участием реакций комплексообразования.
4.3 Равновесие в растворах с участием реакций окисления – восстановления.
4.4Равновесие осадок-раствор.
4.1 Кислотно-оснóвное равновесие
Кислотно-оснóвные реакции – это реакции с переносом протона Н+, поэтому иначе их называют протолитическими реакциями.
В аналитической химии чаще всего используют две теории кислот и оснований:
-теория электролитической диссоциации (теория Аррениуса);
-протолитическая (протонная) теория Бренстеда и Лоури.
Согласно теории Бренстеда и Лоури, вещества, которые участвуют в протолитических реакциях, называются протолитами. Протолиты могут быть молекулярными, катионными и анионными. Типы протолитов приведены в таблице 4.4.1.
Таблица 4.4.1 – Типы протолитов по Бренстеду и Лоури
Тип протолита, определение |
|
Примеры протолитов: |
||
молекулярных |
катионных |
анионных |
||
|
||||
|
|
|
|
|
Кислота – донор Н+ |
HCl |
NH4+ |
HSO4– |
|
Основание – акцептор Н+ |
NH3 |
[Al(OH)(H2O)5]2+ |
HPO42– |
|
Амфолит – вещество, которое может |
H2O |
[Al(OH)(H2O)5]2+ |
H2PO4– |
|
быть и донором, и акцептором Н+ |
||||
|
|
|
||
Протолитическое равновесие – это взаимодействие кислоты и основания с образованием новой кислоты и нового основания. В нем может принимать участие растворитель-амфолит, например, вода.
Сущность протолитического равновесия заключается в обратимом переносе протона Н+ от кислоты к основанию:
В протолитическом равновесии всегда участвуют две сопряженные кислотно-оснóвные пары. В приведенном примере пара 1 состоит из кислоты НА и сопряженного с ней основания А, пара 2 – из кислоты НВ и основания В.
Силу кислотных и оснóвных свойств отдельных компонентов сопряженной пары оценивают с помощью константы кислотности Kа и

687
константы основности Kb.
Константа кислотности – это константа равновесия:
HA + H2O H3O+ + A–
(4.4.1)
Константа основности – это константа равновесия:
B + H2O BH+ + OH–
(4.4.2)
В водном растворе константы кислотности и основности сопря-женной кислотно-оснóвной пары связаны между собой через константу автопротолиза воды KW:
Ka ∙ Kb = KW |
(4.4.3) |
Константа автопротолиза воды KW = 10–14, она характеризует равновесие автопротолиза воды:
H2O + H2O H3O+ + OH–
Если прологарифмировать формулу (4.4.3) с обратным знаком, то получим удобное для использования выражение:
рKa + рKb = 14, |
(4.4.4) |
где рKa = –lgKa, рKb = –lgKb
Таким образом, чем сильнее кислота, тем слабее сопряженное с ней основание и наоборот. Например, НCl – сильная кислота, значит, ион Cl– является чрезвычайно слабым основанием.
Для водных растворов численные значения констант кислотности основности (теория Бренстеда и Лоури) совпадают со значениями констант диссоциации (ионизации) (теория Аррениуса), поэтому можно пользоваться справочной литературой, составленной на основе классической теории. Для молекулярных кислот и оснований константы Kа и Kb, а также их отрицательные логарифмы рKa и рKb приведены в таблицах, для катионных и анионных протолитов эти значения рас-считываются по формулам (4.4.3) или
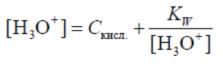
688
(4.4.4).
Равновесия в водных растворах кислот, оснований и амфолитов. Расчет рН протолитических систем:
Водородный показатель рН – это отрицательный логарифм активности или концентрации ионов Н+ (ионов гидроксония Н3О+):
pH = lg aH+
или
pH = lg[H + ] |
(4.4.5) |
Расчет численного значения рН проводят с точностью до сотых долей единицы рН, например, рН = 6,15.
Рассмотрим наиболее распространенные случаи расчета рН.
Равновесия и расчет рН в растворах кислот:
1. Раствор сильной кислоты В растворе сильной кислоты равновесие диссоциации полностью
смещено вправо:
HA + H2O H3O+ + A–
При Скисл > 10–6 М автопротолизом воды можно пренебречь. Тогда равновесная концентрация ионов гидроксония будет равна общей концентрации одноосновной кислоты:
[H3O+] = Скисл. |
|
рН = – lg[H3O+] = – lgСкисл |
(4.4.6) |
Если Скисл ≤ 10–6 М, то надо учесть автопротолиз воды, за счет которого в растворе тоже появляются ионы гидроксония. В этом случае расчет равновесной концентрации ионов гидроксония проводят по формуле:
(4.4.7)
Необходимость учета автопротолиза воды можно продемонстрировать на следующем примере. Рассчитаем значение рН в растворе хлороводородной кислоты при С(HCl) = 10–8 моль/л по формуле (4.4.6), которая используется в теории Аррениуса независимо от концентрации кислоты:

689
рН = – lg10–8 = 8
Ответ является абсурдным (щелочная среда в растворе кислоты!). Если провести расчет по теории Бренстеда-Лоури, т. е. учесть автопротолиз растворителя-амфолита H2O, то по формуле (4.4.7) получим ответ рН = 6,98
2. Раствор слабой кислоты В растворе слабой кислоты происходит неполная диссоциация:
HA + H2O H3O+ + A–
Равновесие характеризуется константой ионизации:
Если выполняются условия:
то можно пользоваться приближенными формулами для расчета рН, сделав допущение, что при указанных условиях равновесная концентрация недиссоциированной кислоты примерно равна ее общей концентрации в растворе:
[HА] ≈ Скисл.
Из уравнения реакции видно, что
Отсюда выражаем равновесную концентрацию ионов гидроксония в растворе слабой кислоты:
После логарифмирования с обратным знаком получаем:

690
(4.4.8)
Если степень диссоциации кислоты α > 5 %, то расчетная формула получается более громоздкой:
(4.4.9)
3. Раствор катионной слабой кислоты Согласно протолитической теории, катионы слабых оснований
являются катионными слабыми кислотами. Рассмотрим расчет значения рН на примере водного раствора хлорида аммония NH4Cl. Эта соль в растворе полностью диссоциирует:
NH4Cl → NH4+ + Cl–
Ион аммония вступает в протолитическую реакцию с растворителемамфолитом водой:
NH4+ + H2O →NH3 + H3O+
В этой реакции аммиак NH3 является молекулярным основанием, ион
– сопряженной катионной кислотой. Следовательно, для расчета рН в растворе NH4Cl выбираем формулу (4.4.8). В нее входит величина рKa, в нашем случае рKa(NH4+), которой в таблицах нет. Для ее расчета используем табличное значение рKb(NH3) и формулу (4.4.4):
рKa(NH4+) = 14 – рKb(NH3)
Подставив это выражение в формулу (4.4.8), получим
(4.4.10)
Раствор многоосновной кислоты В растворе многоосновной кислоты происходит ступенчатая
диссоциация, например, в растворе фосфорной кислоты:
691
В результате образуется сложная многокомпонентная система, и расчет рН сильно усложняется. Так, даже для двухосновной кислоты получается уравнение третьей степени. Поэтому полагают, то при выполнении условия
диссоциация кислоты по второй и третьей ступени подавляется, и рассчитывают значение рН как в растворе слабой одноосновной кислоты, используя формулы (4.8) или (4.9).
Равновесия и расчет рН в растворах оснований:
1. Раствор сильного основания В растворе сильного основания протолитическое равновесие
полностью смещено вправо:
B + H2O BH+ + OH–
При Сосн. > 10–6 М автопротолизом воды можно пренебречь. Тогда равновесная концентрация гидроксид-ионов будет равна общей
концентрации однокислотного основания: |
|
[ОH–] = Сосн. |
|
рОН = – lg[ОH–] = – lgСосн. |
|
рН = 14 – рОН = 14 + lgСосн. |
(4.4.11) |
Если Скисл ≤ 10–6 М, то надо учесть автопротолиз воды, за счет которого в растворе тоже появляются гидроксид-ионы. В этом случае расчет равновесной концентрации ионов ОH– проводят по формуле:
(4.4.12)
2. Раствор слабого основания В растворе слабого основания устанавливается равновесие, которое
смещено влево и характеризуется константой основности:
В+Н2О ↔ ВН++ОН–


693
теории, анионы слабых кислот являются анионными слабыми основаниями. Рассмотрим расчет значения рН на примере водного раствора ацетата натрия CH3COONa. Эта соль в растворе полностью диссоциирует:
CH3COONa CH3COO– + Na+
Ацетат-ион вступает в протолитическую реакцию с растворителемамфолитом водой:
CH3COO– + H2O ↔ CH3COOH + OH–
В этой реакции уксусная кислота CH3COOH является молекулярной кислотой, а ион CH3COO– – сопряженным анионным основанием. Следовательно, для расчета рН в растворе CH3COONa выбираем формулу (4.4.13). В нее входит величина рKb, в нашем случае рKb(CH3COO–), которой в таблицах нет. Для ее расчета используем табличное значение рKа(CH3COOH) и формулу (4.4.4):
рKb(CH3COO–) = 14 – рKа(CH3COOH)
Подставив это выражение в формулу (4.4.13), получим
(4.4.15)
4. Раствор многокислотного основания
Врастворе многокислотного основания происходит ступенчатая диссоциация.
Например, в водном растворе этилендиамина H2NСH2–CH2NH2 образуется гидроксид этилендиаммония, который является двухки-слотным основанием и диссоциирует по ступеням:
Врезультате образуется сложная многокомпонентная система, и расчет рН сильно усложняется. Поэтому полагают, то при выполнении условия

695
ацетатного буфера, который состоит из CH3COOH и CH3COONa. В растворе ацетатного буфера имеют место равновесия:
CH3COOH ↔ CH3COO– + H+
CH3COONa → CH3COO– + Na+
При добавлении к нему небольшого количества сильной кислоты ионы Н+ связываются с ацетат-ионами в молекулы слабой уксусной кислоты, в результате чего значение рН практически не меняется:
CH3COO– + H+ ↔ CH3COOH
При добавлении небольшого количества щелочи ионы ОН– связываются с ионами H+, образуя молекулы воды, поэтому значение рН тоже практически не меняется:
H+ + ОН– ↔ Н2О
В фосфатном буфере, состоящем из двух солей NaH2PO4 и Na2HPO4, оба компонента диссоциированы на ионы:
NaH2PO4 → Na+ + H2PO4–
Na2HPO4 → 2Na+ + HPO42–
При добавлении Н+ гидрофосфат переходит в дигидрофосфат, связывая поступившие протоны:
HPO42– + H+ ↔ H2PO4–
При добавлении ОН– происходит кислотно-основная реакция с участием дигидрофосфата, в ходе которой ионы ОН– связываются в молекулы Н2О:
H2PO4– + ОН– ↔ HPO42– + Н2О
Использование буферных растворов. Буферные растворы используют во всех случаях проведения химических реакций и технологических процессов, когда требуется поддерживать постоянное значение рН в ходе их проведения.
Ход многих аналитических химических реакций, которые используются для обнаружения и разделения, зависит от рН среды, поэтому для поддержания необходимого значения рН используют буферные растворы. В особенности это касается следующих реакций:
1. С участием анионов слабых кислот или катионов слабых оснований.

696
Например, групповые разделения – осаждение катионов II аналитической группы, III аналитической группы с помощью групповых реактивов – проводятся только в аммиачном буфере. Реакции обнаружения ионов Mg2+ c Na2HPO4, PO43– с Mg2+, Ba2+ с K2Cr2O7 и многие другие также проводятся в среде рН-буферов.
2. Цветных качественных реакций с органическими реагентами. Например, при обнаружении ионов Al3+ капельной реакцией с ализарином необходимо, помимо анализируемого раствора и реактива, на-нести на полоску фильтровальной бумаги HCl и поместить ее над ем-костью с конц. NH4OH (первый компонент аммиачного буфера). При взаимодействии кислоты и слабого основания образуется второй компонент буфера – NH4Cl, и тем самым создаются необходимые условия для протекания цветной реакции.
При проведении определения с использованием метода комплексонометрического титрования важнейшим условием также является присутствие буфера в титруемом растворе.
Буферные растворы образуются в ходе кислотно-оснóвного титрования слабых протолитов и их смесей до точки эквивалентности или между двумя точками эквивалентности.
Примеры использования буферов в химической технологии и полиграфии:
-в офсетной печати используется фосфатный буфер с рН = 6,8;
-в гальванопроцессах используется борный буфер с рН 5 и т. д.
В крови человека тоже есть рН-буфер с рН = 7,3–7,5, состоящий из Н2СО3 и НСО3–. Если значение рН крови по каким-либо причинам не укладывается в данный интервал, то это приводит к летальному исходу.
Расчет рН буферных смесей:
1. Буферный раствор на основе слабой кислоты и ее соли Рассмотрим расчет значения рН на примере цианидного буфера,
состоящего из HCN и NaCN. В растворе оба компонента диссоциируют, причем соль полностью:
NaCN → Na+ + CN–,
а кислота – обратимо:
Запишем выражение для константы равновесия:
(4.4.17)

697
Поскольку степень диссоциации (α) слабой кислоты мала, то можно сделать допущение, что равновесная концентрация недиссоциированной кислоты равна ее общей концентрации в растворе:
Кроме того, равновесную концентрацию цианид-ионов можно принять практически равной общей концентрации соли
поскольку NaCN, как сильный электролит, диссоциирует полностью. При этом диссоциация слабой кислоты подавляется за счет поступления ионов CN– из соли.
Подставим эти выражения в формулу (4.4.17) и получим:
(4.4.18)
Используя эту расчетную формулу, можно объяснить также свойства буферных растворов:
-«Значение рН буферного раствора не меняется при разбавлении». Если раствор разбавить в n раз, то концентрация обоих компонентов буфера тоже уменьшится в n раз, однако отношение концентраций останется постоянным значение рН не изменится. Однако, при очень сильном разбавлении ( 104 раз) значение рН меняется на 0,5–1,0 ед. рН.
-«Значение рН буферного раствора мало меняется при добавлении небольших количеств Н+ или ОН–». При добавлении небольшого количества Н+ концентрация слабой кислоты несколько увеличивается, а концентрация соли – уменьшается. При этом отношение концентраций компонентов буфера немного меняется, но логарифм этого отношения меняется очень незначительно значение рН практически не изменяется. Если же добавить к буферу небольшое количество ОН–, то концентрация слабой кислоты несколько уменьшится, а концентрация соли – увеличивается, но логарифм этого отношения меняется очень незначительно значение рН также практически не изменяется.
1. Буферный раствор на основе слабого основания и его соли Рассмотрим расчет значения рН на примере аммиачного буфера,
состоящего из NH4OH и NH4Cl. В растворе оба компонента диссоции-руют, причем соль полностью:
NH4Cl → NH4+ + Cl–

698
а основание – обратимо:
Запишем выражение для константы равновесия:
(4.4.19)
Поскольку степень диссоциации (α) слабого основания мала, то можно сделать допущение, что равновесная концентрация недиссоциированного основания равна его общей концентрации в растворе:
[NH4OH] Cосн.
Кроме того, равновесную концентрацию ионов NH4+ можно при-нять практически равной общей концентрации соли
[NH4+] Cсоли,
поскольку NH4Cl, как сильный электролит, диссоциирует полностью. При этом диссоциация слабого основания подавляется за счет поступления ионов NH4+ из соли.
Подставим эти выражения в формулу (4.4.19) и получим:
(4.4.20)
3. Буферный раствор на основе двух солей многоосновной кислоты Рассмотрим расчет значения рН на примере фосфатного буфера,
состоящего из NaH2PO 4 и Na2HPO4. В этой системе дигидрофосфат натрия NaH2PO4, имеющий больше ионов H+, выступает в роли кислоты по отношению к своей соли – гидрофосфату натрия Na2HPO4 (меньше ионов H+). Следовательно, для расчета значения рН необходимо воспользоваться

699
формулой (4.4.18). Для выяснения вопроса, какое значение pKa фосфорной кислоты надо подставить в формулу, запишем равновесие между компонентами буфера:
Следовательно, необходимо подставить pKa,2. Тогда расчетная формула примет вид:
Буферная емкость. Способность буферных растворов поддерживать значение рН постоянным не безгранична. Любой буферный раствор обладает определенной буферной емкостью.
Буферная емкость – это такое количество моль эквивалентов сильной кислоты или сильного основания, которое нужно добавить к 1 л буферной раствора, чтобы изменить его значение рН на 1 ед.:
(4.4.21)
где n(1/z Х) – количество моль экв. кислоты или щелочи; рН – изменение рН после добавления кислоты или щелочи; V – объем буферного раствора, л.
Чем больше буферная емкость, тем лучше буфер удерживает постоянным значение рН.
Факторы, влияющие на буферную емкость:
-суммарная концентрация компонентов смеси: чем она больше, тем больше буферная емкость;
-соотношение концентраций компонентов буфера: чем ближе к 1 значение
тем больше буферная емкость, поэтому максимальная буферная емкость достигается при одинаковых концентрациях компонентов:
Скисл. = Ссоли или Сосн. = Ссоли
В этих случаях, согласно формулам (4.18) и (4.20) значения рН буферных растворов равны:

700
pH = pKa или pH = 14 – pKb
На практике обычно готовят буферы со значениями рН
pH = pKa ± 1 или pH = (14 – pKb) ± 1
Для этого задают отношения концентраций компонентов буферной
смеси
В интервале от 10 : 1 до 1 : 10. Рецептуры для приготовления буферных растворов с различными значениями рН приводятся в справочнике.
Использование реакций кислотно-основного взаимодействия в аналитической химии. Реакции кислотно-оснóвного взаимодействия широко используются в аналитической химии с различными целями:
-для обнаружения;
-для разделения;
-для определения – кислотно-оснóвное титрование;
-для регулирования значения рН;
-для растворения проб и перевода осадков в раствор.
Константы ионизации – это основные характеристики кислот и оснований (слабых или средней силы), численное значение которых необходимо для решения многих химико-аналитических задач (таблица
4.4.2).
Таблица 4.4.2 – Типы химико-аналитических задач, решаемых с использованием констант ионизации
|
Расчеты с использованием Kа |
|
Расчеты с использованием Kb |
||
|
|
|
Расчет рН в растворах: |
||
- слабых кислот и их солей; |
|
- слабых оснований и их солей; |
|||
- кислот средней силы и их солей; |
|
||||
|
- оснований средней силы и их солей; |
||||
- многоосновных кислот и их солей; |
|||||
- многокислотных оснований и их солей; |
|||||
буферных смесей на основе слабых |
|
- буферных смесей на основе слабых оснований |
|||
- кислот и их солей, кислот средней силы и |
|||||
их |
солей, |
многоосновных кислот |
и их |
и их солей, оснований средней силы и их солей, |
|
многокислотных оснований и их солей |
|||||
солей; амфолитов |
|
||||
|
|
||||
|
|
|
|
||
|
|
Расчет кривых кислотно-основного титрования: |
|||
- |
слабых |
и многоосновных |
кислот |
- слабых и многокислотных оснований сильными |
|
сильными основаниями; |
|
кислотами; |
|||
- солей слабых и многоосновных кислот |
- солей слабых и многокислотных оснований |
||||
сильными кислотами; |
|
сильными основаниями; |
|||
- смесей кислот и смесей кислота + соль |
- смесей оснований и смесей основание + соль |
||||
слабого основания |
|
слабой кислоты |
|||
|
|
|
|
||
|
|
Прогнозирование возможности титрования: |
|||
- слабых кислот и их солей; |
|
- слабых оснований и их солей; |
|||
701
- смесей кислот и смесей кислота + соль |
- смесей оснований и смесей основа-ние + соль |
|
слабого основания |
|
слабой кислоты |
|
|
|
Прогнозирование числа точек эквивалентности и числа скачков на кривых |
||
|
титрования: |
|
|
|
|
- многоосновных кислот и их солей |
|
- многокислотных оснований и их солей |
Расчет ошибок титрования: |
|
|
- кислотной и солевой |
|
- основной и солевой |
4.4.2 Равновесие в растворах с участием реакций комплексообразования
Основные понятия:
Реакции комплексообразования – это реакции с переносом электронных пар и образованием связей по донорно-акцепторному механизму. В них участвуют металл-комплексообразователь (центральный атом, ЦА) и лиганды (L). В результате протекания реакции образуются комплексные соединения (комплексы).
Комплекс – это сложная частица, состоящая из составных частей, способных к автономному существованию.
Состав комплекса:
В состав лиганда входят донорные атомы – атомы, которые связывают лиганд с центральным атомом, например, атомы O, N, S и др.
Комплексообразователь характеризуют координационным числом (КЧ), а лиганд – дентатностью.
Координационное число центрального атома – это число мест во внутренней сфере комплекса, которые могут быть заняты лигандами.
Дентатность лиганда – это число донорных атомов лиганда, ко-торые образуют связи с ЦА.
Моно- и полидентатные лиганды. Хелатные комплексы
В зависимости от количества донорных атомов различают моно- и полидентатные лиганды.
Монодентатные лиганды имеют один донорный атом (H2O, NH3, Cl–, OH–, CN– и др.) и поэтому образуют одну связь с ЦА. Комплексы с монодентатными лигандами имеют нециклическое строение, например, гексафтороферрат (III) [FeF6]3– имеет следующее строение:

702
Полидентатные лиганды имеют несколько донорных атомов, за счет чего образуют несколько связей с ЦА. Комплексы с полидентат-ными лигандами всегда содержат циклы, включающие ЦА. Такие комплексы называются хелатными комплексами (хелатами).
Примеры хелатных комплексов, которые используются в аналитической химии:
1.Ализарин имеет 4 донорных атома кислорода и образует с алюминием хелатный комплекс, в котором центральный атом входит в состав шестичленного цикла:
2.8-оксихинолин имеет два донорных атома (O и N) и образует с алюминием хелатный комплекс, в котором центральный атом входит в состав трех пятичленных циклов:
3.ПАН (структура I) имеет 4 донорных атома и образует с ионами Mn2+
идругими ионами Mе2+ хелатные комплексы с предполагаемой структурой II, в которых ион металла входит в состав четырех пятичленных циклов:

703
Важнейшей особенностью хелатов является их повышенная устойчивость по сравнению с аналогично построенными нециклическими комплексами, поэтому в аналитической химии чаще всего используют полидентатные лиганды и хелатные комплексы.
Увеличение устойчивости комплексов при образовании циклов (по сравнению с аналогично построенными нециклическими комплексами) называется хелатным эффектом.
Факторы, влияющие на устойчивость хелатных комплексов:
- число циклов в хелате: чем больше хелатных циклов в комплексе, тем больше его устойчивость. Например, этилендиаминтетраацетатный комплекс железа (III), имеющий четыре хелатных цикла, более устойчив, чем этилендиаминтетраацетатный комплекс цинка (II) с тремя хелатными циклами:
-размера хелатных циклов: наиболее устойчивы комплексы с пяти-и шестичленными циклами;
-расположения циклов и др.
Комплексные соединения обладают рядом свойств, ценных для химика-аналитика, поэтому широко используются в аналитической химии.
Равновесия в растворах комплексных соединений
Комплексообразование с монодентатными лигандами Комплексообразование с монодентатными лигандами протекает
ступенчато:

704
(4.4.22)
Здесь Kуст. (β) – суммарная константа устойчивости комплекса. Она равна произведению ступенчатых констант устойчивости Ki:
K уст. K1 K 2 |
K 3 ... Kn |
(4.4.23) |
Константа |
устойчивости |
характеризует процесс образования |
комплекса, а константа нестойкости – процесс его диссоциации. Они являются обратными величинами:
(4.4.24)
В аналитической химии принято пользоваться величинами констант устойчивости комплексных соединений.
Реакции ступенчатого комплексообразования с неорганическими монодентатными лигандами чаще всего невозможно использовать в аналитической химии, особенно для целей количественного анализа. Причины этого заключаются в следующем:
-эти реакции идут нестехиометрично, т. к. в растворе всегда присутствует смесь нескольких комплексов;
-некоторые прочные комплексы образуются медленно;
-ступенчатые константы устойчивости большинства комплексов Ki мало различаются между собой, поэтому реакции их образования нельзя использовать для количественного анализа, где должно выполняться требование ΔlgKi 4.
Комплексообразование с полидентатными лигандами В общем виде реакцию комплексообразования иона металла с по-
лидентатным лигандом можно представить следующим образом:

705
Это равновесие характеризуется константой устойчивости:
Реакции комплексообразования с органическими полидентатны-ми лигандами очень широко используются в аналитической химии благодаря следующим особенностям:
- эти реакции идут строго стехиометрично, чаще всего в простом стехиометрическом отношении
M : L=1 : 1
-комплексы образуются в одну стадию (не ступенчато!);
-образующиеся хелатные комплексы очень устойчивы.
Использование реакций комплексообразования в аналитической
химии. Реакции комплексообразования широко используются в аналитической химии с различными целями:
1)для обнаружения;
2)для разделения и маскирования;
3)для определения. На использовании реакций комплексообразования основаны следующие методы количественного химического анализа:
- гравиметрический метод осаждения (если комплексное соединение является малорастворимым);
- комплексометрическое титрование; - косвенные методы определения в кислотно-оснóвном и окис-
лительно-восстановительном титровании;
4)для растворения твердых проб;
5)для изменения кислотно-оснóвных свойств системы. Например,
слабую борную кислоту H3BO3 (pK1 = 9,15; pK2 = 12,74; pK3 = 13,80)
невозможно определить методом кислотно-оснóвного титрования, поскольку при таких значениях pKа скачок на кривой титрования отсутствует. Однако H3BO3 может образовывать более сильные комплексные кислоты с рядом многоатомных спиртов (этиленгликоль, глицерин, маннит и др.). Поэтому до титрования в раствор вводят многоатомный спирт. При этом протекает реакция (на примере этиленгликоля):

706
рKа = 4,48, поэтому при титровании ее раствором щелочи:
Н[В(С2Н4О2)2] + NaOH = Na[В(С2Н4О2)2] + H2O
получается явно выраженный скачок на кривой титрования. Во многих методиках определения борной кислоты используют маннит – шестиатомный спирт, изомер сорбита;
6) для изменения окислительно-восстановительных свойств системы. Как показано в разделе 4, связывание в комплекс окисленной или восстановленной форм всегда вызывает изменение потенциала.
Константа устойчивости Kуст. – основная характеристика комплексного соединения, численное значение которой необходимо для решения многих химико-аналитических задач:
1)расчет равновесных концентраций ионов в растворах комплексных соединений;
2)расчет сложных равновесий в многокомпонентных растворах:
-расчет растворимости малорастворимого электролита, если один из ионов одновременно участвует в реакции комплексообразования;
-расчет окислительно-восстановительного потенциала, если один из компонентов сопряженной окислительно-восстановительной пары одновременно участвует в реакции комплексообразования;
3) расчет кривых комплексометрического титрования;
4) выбор маскирующего агента для конкретного иона;
5) оценка возможности обнаружения или количественного определения конкретного иона с использованием реакций комплексообразования;
6)прогнозирование возможности титрования ионов-комплексообра- зователей или ионов-лигандов;
7)расчет индикаторных ошибок комплексонометрического титрования (по значениям Kуст. комплексонатов металлов).
4.4.3 Равновесие в растворах с участием реакций окислениявосстановления
Окислительно-восстановительные реакции (ОВР) – это реакции с переносом электрона ē.

707
Обратимую окислительно-восстановительную полуреакцию можно записать в общем виде:
Для характеристики ОВР удобнее использовать не константу равновесия, а электродный потенциал Е. Согласно уравнению Нернста:
(4.4.25)
В это уравнение входят следующие величины: Е – равновесный электродный потенциал, В;
Е0 – стандартный электродный потенциал, В (табличная величина); R – универсальная газовая постоянная, R = 8,314 Дж/(моль∙К);
Т – абсолютная температура, К;
n – число электронов в полуреакции; F – число Фарадея, F = 96500 Кл/моль;
ао.ф. и ав.ф. – активности окисленной и восстановленной форм.
Правила записи уравнения Нернста. Уравнение Нернста составляют для полуреакции, в которой участвуют компоненты сопряженной окислительно-восстановительной пары, используя следующие правила:
1. Если в полуреакции участвуют
-твердые фазы,
-металлы,
-газы при р = 1 атм.,
-растворители,
то их можно исключить из уравнения Нерста, поскольку их активность принимают равной а = 1.
2.Активности веществ возводят в степени, равные стехиометрическим коэффициентам.
3.Для разбавленных растворов:
поэтому можно заменить активности на концентрации:
(4.4.26)
4. Часто все постоянные величины объединяют в одну константу и заменяют натуральный логарифм десятичным. Так, при 25 С уравнение имеет вид

708
(4.4.27)
При 20 ºС предлогарифмический множитель равен 0,058/n, а при 30 С –
0,060/n.
5. Если в полуреакции участвуют ионы Н+, ОН– или какие-либо другие, то их концентрацию включают в уравнение Нернста в соответствующей степени.
Факторы, влияющие на величину электродного потенциала:
На величину электродного потенциала Е влияют следующие факторы:
1)Химическая природа компонентов пары: Е0 и n.
2)Концентрации окисленной и восстановленной форм: чем больше Cок.ф., тем больше значение E; чем больше Cв.ф., тем меньше E.
3)Ионная сила раствора: ионная сила I влияет на величины коэффициентов активности γ, поэтому с изменением I изменяются активности ионов в растворе. Поскольку заряды окисленной и восстановленной форм обычно различны, то их активности меняются в различной степени. Если отношение активностей аок.ф./ав.ф. увеличивается, то значение E тоже увеличивается и наоборот.
4)Значение рН раствора: величина рН влияет на Е явно, если в уравнение Нернста входят концентрации ионов H+ или OH–.
5)Другие побочные реакции, в которые вступают окисленная или восстановленная формы (реакции осаждения, комплексообразования): если в осадок или комплекс связывается окисленная форма, то величина электродного потенциала уменьшается, а если связывается вос-становленная форма, то Е увеличивается.
Направление окислительно-восстановительных реакций:
ОВР протекает в заданном направлении, если ЭДС реакции (ΔЕ)
∆E=Еок–Евос>0
Если Е < 0, то ОВР протекает в обратном направлении, а если Е = 0, то наблюдается состояние равновесия. Чем больше Е, тем интенсивнее, быстрее протекает ОВР.
Минимальное значение Е для аналитической ОВР должно составлять
0,2–0,3 В.
Количественной характеристикой направления и полноты протекания ОВР является константа равновесия K:
(4.4.28)

709
где Е0ок – стандартный электродный потенциал окислителя, В;
– стандартный электродный потенциал предполагаемого восстановителя, В;
n – число электронов, участвующих в ОВР (наименьшее общее кратное числа электронов, участвующих в полуреакциях).
Чем больше величина K, тем полнее протекает ОВР.
Использование реакций окисления-восстановления в аналитической химии. Реакции окисления-восстановления широко используются в аналитической химии с различными целями:
1)для обнаружения;
2)для разделения;
3)для определения. На использовании ОВР основаны следующие методы количественного химического анализа:
-гравиметрический метод выделения;
-методы окислительно-восстановительного титрования;
-косвенные методы определения в кислотно-основном и комплексонометрическом титровании;
4) для растворения или разложения твердых проб.
При выполнении анализа на основе ОВР проводят различного типа расчеты и обоснования с использованием табличной величины Е0. Численное значение Е0 необходимо для решения многих химико-аналитических задач:
1)определение направления ОВР для выяснения возможности окисления или восстановления анализируемого вещества конкретным аналитическим реагентом с целью обнаружения или определения;
2)расчет константы равновесия ОВР (K) для выяснения полноты протекания аналитической реакции;
3)выбор подходящего окислителя или восстановителя на основании сравнения Е10 и Е20 или расчета K;
4)прогнозирование возможности применения данной ОВР для проведения титриметрического определения;
5)расчет индикаторных ошибок титрования;
6)химико-аналитические расчеты по уравнению Нернста:
-расчет кривых окислительно-восстановительного титрования;
-расчет условий проведения анализа с использованием ОВР – значения рН, концентрации реагирующих веществ, концентрации маскирующих агентов или реагентов-осадителей (учет побочных реакций), в т. ч. с целью увеличения скачка кривой титрования.
4.4.4 Равновесие осадок-раствор
Между малорастворимым соединением (МРС) и его ионами в растворе устанавливается равновесие (заряды ионов опущены):
710
Константа равновесия этой реакции называется константой растворимости или произведением растворимости (KS или ПР).
Способы выражения константы растворимости:
- термодинамическая константа растворимости KS0 записывается через активности ионов:
KS0 = аAm • aBn
(4.4.29)
Эта величина приводится в справочниках, она является константой при данных температуре, давлении и природе растворителя.
- реальная (концентрационная) константа растворимости KS записывается через равновесные концентрации ионов:
KS = [A]m • [B]n (4.4.30)
Связь между термодинамической и реальной константами растворимости:
KS 0=аAm • a Bn = γ mA  γ nB • [ A]m • [ B ]n= γ mA • γnB •KS
γ nB • [ A]m • [ B ]n= γ mA • γnB •KS
(4.4.31)
Растворимость осадков и факторы, влияющие на нее
Растворимость – это общая концентрация вещества в его насыщенном растворе.
Растворимость МРС вычисляется из табличного значения KS0:
-без учета коэффициентов активности γ, если ионная сила раствора I 0. Так рассчитывают, например, растворимость МРС в воде при очень низких значениях KS0 (концентрация растворенной части МРС не превышает 10–4 моль/л);
-с учетом коэффициентов активности γ, если ионная сила раствора I >
0.Это бывает в случае заметной растворимости МРС (высокое значение KS0) или присутствия в растворе посторонних сильных электролитов, которые создают ионную силу.
В аналитической химии осаждение считается практически полным, если остаточная концентрация осаждаемого иона в растворе
Сост. ≤ 10–6 моль/л Факторы, влияющие на растворимость осадков:
1) Природа осадка и растворителя. Осадки, состоящие из неорганических ионов, лучше растворяются в воде, чем в органических растворителях. Осадки, содержащие крупные органические ионы, лучше растворяются в органических растворителях. Поэтому для увеличения или уменьшения растворимости в раствор вводят органические растворители.
712
Причины понижения растворимости: концентрации ионов осадка взаимосвязаны через величину константы растворимости. Следовательно, при увеличении концентрации одного иона концентрация другого понижается, поскольку KS = const:
Ks = [A]m • [B]n
Иначе, одноименные ионы смещают равновесие в сторону процесса осаждения, поэтому растворимость уменьшается. Например, при введении одноименных ионов В равновесие, которое установилось в насыщенном растворе МРС, смещается влево:
AmBn = mA + nB
Таким образом, для того, чтобы сделать осаждение более полным, надо брать 1,5-кратный избыток осадителя.
Причины увеличения растворимости:
1)Солевой эффект. При большом избытке одноименных ионов возрастает ионная сила раствора, в результате чего влияние I становится сильнее действия одноименного иона.
2)Химические взаимодействия осадка с избытком осадителя:
- комплексообразование, например
HgI2 + 2I– [HgI4]2–
- растворение амфотерных гидроксидов в избытке щелочи:
Zn(OH)2 + 2OH– [Zn(OH)4]2–
- образование более растворимых кислых солей:
CaCO3 + H2O + CO2 Ca(HCO3)2
Влияние конкурирующих реакций на растворимость МРС. Если катион или анион МРС вступает в конкурирующую реакцию, то растворимость увеличивается вплоть до полного растворения осадка. Для растворения осадков используют реакции всех типов:
1) Кислотно-оснóвные реакции:
- осадки, которые содержат анионы слабых кислот, растворяются в кислотах, например,
BaCO3 + 2H+ → Ba2+ + H2O +CO2↑
- осадки, которые содержат катионы амфотерных гидроксидов, растворяются в щелочах, например,
PbSO4 + 6OH– → [Pb(OH)6]4– + SO42–
714
аналитической группы. Сначала к пробе добавляют NH4ОН (один из компонентов буферного раствора с рН = 9,0–9,2). При этом начинает проходить побочная реакция – осаждение катиона I группы Mg2+ в виде гидроксида:
Mg2+ + 2NH4ОН→Mg(OH)2 + 2NH4+
Далее по методике добавляют раствор NH4Cl (второй компонент буфера). При этом значение pH раствора понижается, т. к. вместо слабого основания в растворе появляется буфер. В результате понижения pH гидроксид магния растворяется и не мешает осаждению катионов II группы под действием (NH4)2CO3.
При разделении катионов III аналитической группы внутри группы также используют смещение равновесия осаждения-растворения.
В ходе разделения сначала все ионы вступают в реакцию со щелочью, образуя нерастворимые гидроксиды:
Fe2+ + 2OH– = Fe(OH)2↓ |
Cr3+ |
+ 3OH– = Cr(OH)3↓ |
Fe3+ + 3OH– = Fe(OH)3↓ |
Zn2+ |
+ 2OH– = Zn(OH)2↓ |
Mn2+ + 2OH– = Mn(OH)2↓ |
Al3+ |
+ 3OH– = Al(OH)3↓ |
Затем при добавлении избытка щелочи амфотерные гидроксиды растворяются:
Cr(OH)3↓ + 3OH– = [Cr(OH)6]3–
Zn(OH)2↓ + 2OH– = [Zn(OH)4]2–
Al(OH)3↓ + 3OH– = [Al(OH)6]3–
В результате часть катионов остается в растворе, а часть находится в осадке в виде малорастворимых гидроксидов.
Использование реакций осаждения в аналитической химии.
Реакции осаждения широко используются в аналитической химии с различными целями:
1)для обнаружения;
2)для разделения;
3)для определения. На использовании реакций осаждения основа-ны следующие методы количественного химического анализа:
-гравиметрический метод осаждения;
-осадительное титрование;
-косвенные методы определения в кислотно-оснóвном, окислительновосстановительном и комплексометрическом титровании.
При выполнении анализа с использованием реакций осаждения возникает необходимость в проведении различного типа расчетов и обоснований с использованием табличной величины KS0. Константа растворимости – одна из основных характеристик осадка, численное
715
значение которой необходимо для решения многих химико-аналитических задач:
1) расчет растворимости осадков при заданных условиях: - в воде (через KS0 при I → 0 или через KS при I > 0);
- в присутствии одноименных ионов (через KS0 при I → 0 или через KS
при I > 0);
- в присутствии разноименных ионов (через KS);
2) расчет условий растворения и осаждения осадков:
-условий количественного осаждения малорастворимого соединения;
-условий начала образования осадка;
-условий, при которых осадок не выпадает;
3)прогнозирование возможности выпадения осадка при смешении растворов заданной концентрации (путем сравнения KS0 и ПС);
4)выбор осадителя и осаждаемой формы для конкретного иона;
5)оценка возможности обнаружения или количественного определения конкретного иона с использованием реакций осаждения.

716
4.5 МЕТОДЫ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
ПЛАН
4.5.1Общие сведения о разделении и концентрировании.
4.5.2Классификация методов разделения и концентрирования.
4.5.3Экстракция как метод разделения и концентрирования.
4.5.4Сорбция как метод разделения и концентрирования.
4.5.5Ионный обмен.
4.5.6Методы осаждения и соосаждения.
4.5.7Электролитическое выделение и цементация.
5.5.8 Методы испарения.
4.5.9 Количественные характеристики концентрирования.
4.5.1 Общие сведения о разделении и концентрировании
Разделение – это операция, позволяющая отделить компоненты пробы друг от друга.
Его используют, если одни компоненты пробы мешают определению или обнаружению других, т. е. когда метод анализа недостаточно селективен и надо избежать наложения аналитических сигналов. При этом обычно концентрации разделяемых веществ близки.
Концентрирование – это операция, позволяющая увеличить концентрацию микрокомпонента относительно основных компонентов пробы (матрицы).
Его используют, если концентрация микрокомпонента меньше предела обнаружения Сmin, т. е. когда метод анализа недостаточно чувствителен. При этом концентрации компонентов сильно различаются. Часто концентрирование совмещается с разделением.
Виды концентрирования:
1. Абсолютное: микрокомпонент переводят из большого объёма или большой массы пробы (Vпр или mпр) в меньший объём или меньшую массу концентрата (Vконц или mконц). В результате концентрация микрокомпонента увеличивается в n раз:
n Vпр Сконц ,
Vконц Спр
(4.5.1)
где n – степень концентрирования.
Чем меньше объём концентрата, тем больше степень концентрирования. Например, 50 мг катионита поглотили германий из 20 л водопроводной воды, затем германий десорбировали 5 мл кислоты. Следовательно, степень концентрирования германия составила:

|
717 |
||
n |
20000 мл |
4000 раз . |
|
5 мл |
|||
|
|
||
2. Относительное (обогащение): микрокомпонент отделяется от макрокомпонента так, что отношение их концентраций увеличивается. Например, в исходной пробе отношение концентраций микро- и макрокомпонентов составляло 1:1000, а после обогащения – 1:10. Обычно это достигается путём частичного удаления матрицы.
Разделение и концентрирование имеют много общего, для этих целей используются одни и те же методы. Они очень разнообразны. Далее будут рассмотрены методы разделения и концентрирования, имеющие наибольшее значение в аналитической химии.
4.5.2 Классификация методов разделения и концентрирования
Существует множество классификаций методов разделения и концентрирования, основанных на разных признаках. Рассмотрим важнейшие из них.
Классификация по природе процесса дана на рисунке 4.5.1.
Рисунок 4.5.1 Классификация методов разделения по природе процесса
Химические методы разделения и концентрирования основаны на протекании химической реакции, которая сопровождается осаждением продукта, выделением газа. Например, в органическом анализе основным методом концентрирования является отгонка: при термическом разложении матрица отгоняется в виде СО2 , Н2О, N2 , а в оставшейся золе можно определять металлы.
Физико-химические методы разделения и концентрирования чаще всего основаны на избирательном распределении вещества между двумя фазами. Например, в нефтехимической промышленности наибольшее значение имеет хроматография.
Физические методы разделения и концентрирования чаще всего основаны на изменении агрегатного состояния вещества.

718
Классификация по физической природе двух фаз представлена на рисунке 4.5.2. Распределение вещества может осуществляться между фазами, которые находятся в одинаковом или разном агрегатном состоянии: газообразном (Г), жидком (Ж), твёрдом (Т). В соответствии с этим различают следующие методы (рис.).
Рисунок 4.5.2 – Классификация методов разделения по природе фаз
В аналитической химии наибольшее значение нашли методы разделения и концентрирования, которые основаны на распределении вещества между жидкой и твёрдой фазой.
Классификация по количеству элементарных актов (ступеней):
1.Одноступенчатые методы – основаны на однократном распределении вещества между двумя фазами. Разделение проходит в статических условиях.
2.Многоступенчатые методы – основаны на многократном распределении вещества между двумя фазами. Различают две группы многоступенчатых методов:
- с повторением процесса однократного распределения (например, повторная экстракция). Разделение проходит в статических условиях;
- методы, основанные на движении одной фазы относительно другой (например, хроматография). Разделение проходит в динамических условиях.
Классификация по виду равновесия (рисунок 4.5.3).

719
Рисунок 4.5.3 – Классификация методов разделения по виду равновесия
Термодинамические методы разделения основаны на различии в поведении веществ в равновесном состоянии. Они имеют наибольшее значение в аналитической химии.
Кинетические методы разделения основаны на различии в поведении веществ во время процесса, ведущего к равновесному состоянию. Например, в биохимических исследованиях наибольшее значение имеет электрофорез. Остальные кинетические методы используются для разделения частиц коллоидных растворов и растворов высокомолекулярных соединений. В аналитической химии эти методы применяются реже.
Хроматографические методы основаны и на термодинамическом, и на кинетическом равновесии. Они имеют огромное значение в аналитической химии, поскольку позволяют провести разделение и одновременно качественный и количественный анализ многокомпонентных смесей.
4.5.3 Экстракция как метод разделения и концентрирования
Экстракция – наиболее распространенный метод разделения и концентрирования.
Экстракция – это метод разделения, выделения и концентрирования веществ, основанный на распределении растворенного веще-ства между двумя несмешивающимися фазами (в основном жидки-ми), т.е. это процесс распределения вещества между двумя несме-шивающимися жидкостями.
Наибольшее распространение имеют системы, в которых одной фазой является вода, второй – органический растворитель.
Экстракция – процесс сложный, он базируется на химии растворов, комплексных соединений и др.
Через границу раздела фаз проходят не ионы, а нейтральные молекулы. Поэтому вещество должно:
-сольватироваться (взаимодействовать с растворителем);
-обладать лиофобными свойствами (не должно содержать лиофильных групп);
-иметь большие размеры;
-быть устойчивым в водной фазе.
720
Основные понятия:
1.Экстрагент – смесь из органического реагента и растворителя.
2.Органический реагент – вещество, которое непосредственно экстрагирует компонент.
3.Экстракт – органическая фаза, содержащая органический реагент и выделяемый компонент.
4.Экстрагирующееся соединение – вещество, которое переходит органическую фазу.
5.Реэкстракция – процесс, противоположный экстракции (извлечение вещества из экстракта в водную фазу).
Классификация экстракционных методов. По технике выполнения:
1.Периодическая экстракция (однократная, двухкратная). Используется, когда коэффициенты распределения велики.
Проводится в делительных воронках, когда экстрагент тяжелее воды (находится на дне воронки) или, наоборот, экстрагент легче воды (находится на поверхности).
2.Непрерывная экстракция. Используется при небольших значениях коэффициентов распределения.
Если растворитель (экстрагент) легче воды, тогда он первым вносится на дно сосуда, а затем, поднимаясь, экстрагирует вещества. Если растворитель тяжелее воды, то он капает из воронки через воду собирается на дне сосуда.
3.Способ фракционирования. В этом случае прибор для экстракции состоит из ряда сосудов.
4.Колоночная экстракция (хроматографическая). Неподвижная фаза наносится на инертный носитель, через нее пропускается подвижная фаза.
По характеристике экстрагента:
1.Кислые экстрагенты.
2.Основные экстрагенты.
3.Нейтральные экстрагенты (кетоны, спирты, эфиры).
По природе экстрагирующегося вещества:
1.Координационно несольватированные нейтральные соединения с ковалентными связями (координационно насыщенные простые соединения),
например, AsI3, HgCl2, OsO4, SbI3 и др.
2.Хелаты (внутрикомплексные соединения), образуемые иона-ми металлов с реагентами, которые содержат по крайней мере два атома (O, N, S
идр.), способных одновременно координироваться с металлом. Это самый распространенный класс экстрагирующихся соединений.
3.Координационно несольватированные соли (координационно насыщенные соединения). Это комплексные катионы, извлекаемые в присутствии подходящих противоионов. Сюда же относятся и ионные ассоциаты. Например, FeL3, где L – 1,10-фенентролин, противо-ион – перхлорат.
721
4.Минеральные кислоты (HCl, HNO3, H2SO4 и др.), извлекаемые полярными растворителями с высокой основностью (спирты, эфиры, кетоны
идр.).
5.Комплексные металлокислоты общей формулы Hp-qMXp, где (p-q) обычно равно 1 или 2, например, HFeCl4 или H2CdI4. Эти соединения хорошо экстрагируются активными растворителями, способными к протонизации в кислой среде.
6.Координационно сольватированные нейтральные соединения. Во внутреннюю координационную сферу атома металла входят как неорганические анионы, содержащиеся в водной фаза, так и молекулы экстрагента. Эти соединения извлекаются высокоактивными растворителями, способными к координации с металлом.
Характеристика растворителей. При выборе растворителя необходимо учитывать его свойства, которые обеспечивают взаимодействие растворителя с растворенным веществом.
1.Растворитель и растворенное вещество должны быть близки по свойствам.
2.Необходимо учитывать ε – диэлектрическую проницаемость (процессы диссоциации и ионизации). Если ε растворителя больше 40, то вещество находится в виде ионов.
3.Очень важным является сольватация. Сольватация – процесс неизбирательный. Сольватация бывает конечной, когда молекулы сольвата занимают строго определенные места (например, гидраты), вторичная, когда имеется вторичный слой растворителя с неопределенным количеством сольватных молекул.
4.Ориентационное взаимодействие между молекулами растворителя также оказывает влияние на экстракцию.
5.Необходимо учитывать возможное образование водородных связей. На практике к растворителям предъявляются следующие требования:
1.Растворитель не должен смешиваться с водой.
2.Плотность растворителя должна существенно отличаться от плотности воды.
3.При повторных экстракциях предпочитают растворители тяжелее
воды.
4.Температура кипения растворителя должна быть оптимальной. Например, испарение мешает работе, так как изменяется соотношение объемов фаз.
5.Растворитель не должен образовывать эмульсии.
6.Растворитель не должен разлагаться.
Достоинства экстракции:
1.Метод пригоден для абсолютного и относительного концентрирования.
2.Используется для группового и индивидуального выделения элементов при анализе природных и промышленных объектов.
722
3.Универсальность по отношению к природе объектов и их концентраций.
4.Совмещаемость высокой эффективности концентрирования с разнообразными методами определения.
Сочетание экстракции с методами определения. Экстракционно-
фотометрические методы. Производят измерение оптической плотности экстракта. Главное достоинство – увеличение селективности.
Экстракционно-люминесцентные методы. Измеряют люминесценцию экстракта, растворитель при этом не должен люминесцировать. Главное достоинство – увеличение чувствительности и селективности.
Экстракция и атомно-абсорбционные методы. Увеличивается чувствительность в пламенном варианте и избирательность удаления матрицы.
Экстракция и атомно-эмиссионный спектральный анализ (эмиссионная фотометрия пламени). Упрощается градуировка за счет устранения матрицы, увеличивается чувствительность, снижается погрешность. Но при этом часто снижается число определяемых элементов.
Экстракция и вольтамперометрия. Производится полярографирование экстрактов после введения элемента и растворителя. Можно определять гидролизующиеся элементы.
4.5.4 Сорбция как метод разделения и концентрирования
Сорбция – это поглощение газов или растворённых веществ твёрдыми или жидкими сорбентами.
Поглощение веществ может происходить на поверхности фазы (адсорбция) или в объёме фазы (абсорбция). В аналитической химии чаще всего применяют адсорбцию с целью:
-разделения веществ, если создать условия для селективного поглощения;
-концентрирования (реже).
Кроме того, сорбция в динамических условиях положена в основу важнейшего метода разделения и анализа – хроматографии.
Классификация:
-По механизму.
-По способу осуществления процесса.
Механизмы сорбции:
1. Адсорбция – поглощение вещества на поверхности твердого или
жидкого тела.
2.Абсорбция – поглощение газов, паров или растворенных веществ во всем объеме твердой или жидкой фазы.
3.Хемосорбция – поглощение веществ твердыми или жидкими сорбентами с образованием химических соединений.
4.Капиллярная конденсация – образование жидкой фазы в порах капиллярах твердого сорбента при поглощении паров вещества.

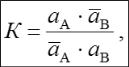
724
(4.5.3)
Где, а – активности ионов.
Если К > 1, то ион В обладает бóльшим сродством к иониту; если К < 1, то ион А обладает бóльшим сродством к иониту; если же К ≈ 1, то оба иона одинаково сорбируются ионитом.
На протекание ионного обмена влияют следующие факторы:
1)природа ионита;
2)природа иона: чем больше отношение заряда иона к радиусу гидратированного иона (z/r), тем больше сродство к иониту;
3)свойства раствора:
-значение рН;
-концентрация иона: из разбавленных растворов ионит сорбирует ионы
сбóльшим зарядом, а из концентрированных – с меньшим;
-ионная сила раствора: чем меньше μ, тем лучше сорбируются ионы. Виды ионитов. Существует большое количество самых разнообразных
ионитов. Они классифицируются по происхождению и по знаку заряда обменивающихся ионов.
Взависимости от происхождения различают две группы ионитов:
1. Природные иониты:
- неорганические (глины, цеолиты, апатиты); - органические (целлюлоза).
2. Синтетические иониты:
- неорганические (пермутиты); - органические (высокомолекулярные материалы).
Ваналитической химии чаще всего используются синтетические органические иониты.
Взависимости от знака заряда обменивающихся ионов иониты называются следующим образом:
1. Катиониты – обменивают катионы, содержат кислотные группы:
1) –SO3H (сильнокислотные катиониты, обмен происходит в широком интервале значений рН);
2) –РO3H2 (среднекислотные катиониты, обмен происходит при рН >
4);
3) –СООН, –ОН (слабокислотные катиониты, обмен происходит при рН > 5).
2. Аниониты – обменивают анионы, содержат оснóвные группы:
|
|
|
|
- |
четвертичные алкиламмониевые группы N R 3 (высокооснóвные |
||
аниониты, обмен происходит в широком интервале значений рН); |
|||
|
|
|
|
- |
амино– и иминогруппы N Н3 , |
N Н2 , |
N Н (средне– и |
низкооснóвные аниониты, обмен происходит при рН < 8–9).
726
4. Вещества, выделяемые в элементном состоянии (ртуть, теллур, селен, золото).
Например, в сильнокислотной (HNO3) среде можно отделить кремниевую, танталовую, ниобиевую и вольфрамовую кислоты практически от всех элементов. Применение разбавленных растворов серной кислоты (иногда в смеси с этанолом) позволяет отделить в виде сульфатов ионы бария, стронция, кальция и свинца от ионов всех элементов.
С помощью фениларсоновой кислоты в кислых растворах можно отделить цирконий(IV) от ионов многих элементов. Широко используют методы осаждения с маскированием. Например, ионы Fe(III), Al(III), Ga(III), Ti(IV), Zr(IV), Th(IV) под действием аммиака осаждаются в виде гидроксидов, тогда как ионы Co(II), Ni(II), Zn(II), Cu(II), Cd(II), образующие с аммиаком комплексы, остаются в растворе.
Соосаждение. Это распределение микрокомпонента между раствором (жидкой фазой) и осадком (твердой фазой), причем микрокомпонент не образует в данных условиях собственной твердой фазы.
Например, если на раствор, содержащий смесь ВаСl2 с FeCl3, подействовать серной кислотой, то следовало бы ожидать, что будет осаждаться только BaSO4, так как соль Fe2(SO4)3 растворима в воде. Однако в действительности вместе с BaSO4 частично осаждается и Fe(III), в чем можно убедиться, прокалив отфильтрованный осадок: остаток оказывается не чисто белым (BaSO4), а окрашенным в коричневатый цвет из-за присутствия оксида железа, образовавшегося при прокаливании:
Fe2(S04)3 = Fe2O3 + 3SO3.
При соосаждении имеют место адсорбция, ионный обмен, окклюзия, изоморфное соосаждение, образование химических соединений и другие виды взаимодействия микрокомпонентов с компонентами осадка. На соосаждение микрокомпонентов оказывают влияние состояние микрокомпонента в растворе, кристаллохимические свойства (структура, поверхность и др.) осадка, процесс старения осадка, кислотность раствора, порядок добавления реагентов, температура, время и другие факторы.
При адсорбции загрязняющее вещество находится на поверхности твердой фазы, которая называется в этом случае адсорбентом.
При окклюзии соосажденные примеси находятся внутри частиц осадка вследствие образования химических соединений между осадком и соосаждаемой примесью, или вследствие явления внутренней адсорбции в процессе формирования осадка, или же вследствие изоморфизма (образование смешанных кристаллов ионами с одинаковым координационным числом и близкими радиусами). В приведенном примере соосаждение играет отрицательную роль и приводит к погрешностям анализа, т. е. к получению неправильных результатов.
Однако соосаждение может быть также полезным как метод
727
концентрирования микрокомпонентов. В аналитической практике нередки случаи, когда концентрация определяемого компонента в растворе настолько мала, что осаждение в его простой форме обычно нельзя применить, даже если осадок имеет очень низкую растворимость (так как при такой концентрации не достигается ПР соответствующего осадка); может также образоваться коллоидный раствор, из которого выделить твердую фазу (осадок) затруднительно. Именно в таких случаях полезно применять соосаждение определяемого микрокомпонента пробы с каким-либо подходящим коллектором.
Коллекторы (носители) – это малорастворимые неорганические или органические соединения, которые должны полностью захватывать нужные и не захватывать мешающие микрокомпоненты и компоненты матрицы. В качестве неорганических коллекторов используют гидроксиды, сульфиды, фосфаты, т. е. преимущественно соединения, образующие аморфные осадки с большой активной поверхностью.
Среди органических коллекторов различают в основном три вида:
1)малорастворимые ассоциаты, состоящие из объемного органического катиона и аниона (например, катион кристаллического фиолетового или метиленового синего и тиоцианат-анион или иодид-анион);
2)хелаты (дитиокарбаминаты, дитизонаты и т. п.);
3)индифферентные органические соединения, которые не содержат комплексообразующих группировок.
Для коллекторов первого вида соосаждаемый элемент, как правило, входит в состав комплексного аниона, например, M(SCN)m- или MIm-. При использовании индифферентных коллекторов в раствор вводят органический реагент, в молекуле которого содержится характерная функциональноаналитическая группировка на определенный микрокомпонент или группу микрокомпонентов, при этом образуется соединение, которое захватывается коллектором. В основном микроэлементы соосаждаются в виде хелатов и ионных ассоциатов.
Механизм соосаждения на неорганических и органических коллекторах различен. В случае неорганического коллектора распределение обусловлено прежде всего его ионной природой и неравномерным распределением зарядов по поверхности из-за наличия поверхностных дефектов. Органические коллекторы образуют молекулярную решетку, на которой ионы практически не сорбируются. Эти различия между коллекторами объясняют высокую избирательность органических коллекторов. Например, гидрохлоридом индулина из хлоридных растворов, содержащих ионы галлия
иалюминия в соотношении 1:8·109, можно выделить ~ 90% галлия; при этом ионы алюминия практически не захватываются.
Эффективность органических коллекторов настолько высока, что селективное выделение микрокомпонента в некоторых случаях осуществляется, когда его отношение к макрокомпоненту может составлять 1:1015. Преимущество органических коллекторов состоит также в простоте
728
обработки: из концентрата можно легко выжечь органическое вещество, концентрат удобно растворить в органическом растворителе.
Соосаждение микрокомпонентов с коллектором выполняют одним из следующих способов:
-введением макрокомпонента и подходящего для него реагентаосадителя (как неорганического, так и органического);
-частичным осаждением матрицы (макрокомпонент присутствует в
пробе);
-введением органического соединения в органическом растворителе, смешивающемся с водой (после разбавления водой соосадитель выпадает в осадок, увлекая за собой комплексы микроэлементов).
4.5.7 Электролитическое выделение и цементация
Наиболее распространен метод электролитического выделения, при котором отделяемое или концентрируемое вещество выделяют на твердых электродах в элементном состоянии или в виде какого-либо соединения. Электролитическое выделение (электролиз) основано на осаждении вещества электрическим током при контролируемом потенциале. Наиболее распространен вариант катодного осаждения металлов; анодное осаждение, например, в форме оксида PbO2, используют редко. Материалом электродов могут служить углерод (графит, стеклоуглерод), платина, серебро, медь, вольфрам, сплавы платины с иридием. Часто выделение проводят на ртутном микрокатоде.
Состав выделяемого соединения зависит от условий электровыделения, свойств компонентов и материала электрода. Например, на графитовом электроде при потенциалах 5–40 мВ некоторые элементы (Ag, Bi, Cd, Cu, Pb) выделяются в элементном состоянии, часть в виде оксидов (Co, Cr, Fe, Mn), гидроксидов или сплавов элементов (Ba, Са, Mg, Mo, Ti, V). Если платина – катод, то выделяются металлы Ni, Ag, Bi, Cd, Co, Pb, Tl, если анод – оксиды Со, Pb, Tl, Ni.
Электролитическое выделение в большинстве случаев составляет неотъемлемую стадию инверсионных методов электроаналитической химии, из которых наиболее распространена инверсионная вольтамперометрия. В случае инверсионных электроаналитических методов анализа стадию предварительного электролитического выделения сочетают с последующими электрохимическими превращениями концентрата, выделенного на ртутных или твердых электродах. Определение заключается в электролитическом растворении ранее выделенного на поверхности электрода вещества. Поскольку концентрация определяемого вещества на стационарном электроде во много раз выше, чем в первоначальном растворе, ток, протекающий при растворении, значительно выше максимального тока до концентрирования. Этим методом определяют концентрации веществ в интервале 10–8 – 10–7 моль/л.
Метод цементации (называемый также внутренним электролизом)
729
заключается в восстановлении компонентов (обычно малых количеств) на металлах с достаточно отрицательными потенциалами (алюминий, цинк, магний) или на амальгамах электроотрицательных металлов без наложения тока. При цементации происходят одновременно два процесса: катодный (выделение компонента) и анодный (растворение цементирующего металла). В качестве примера можно привести выделение микроэлементов из вод на металлах-цементаторах (Al, Mg, Zn), обладающих простыми эмиссионными спектрами, поэтому последующее атомно-эмиссионное определение микроэлементов непосредственно в концентрате легко осуществляется.
4.5 Методы испарения
Методы испарения основаны на разной летучести матрицы и микрокомпонентов. Эти методы применяют при анализе различных объектов; главные их достоинства – простота и доступность. Различают простую отгонку (выпаривание), ректификацию, молекулярную дистилляцию (дистилляцию в вакууме). Важное место занимает отгонка в результате химических превращений – метод, основанный на переводе нелетучих форм микроили макроколичеств элементов или органических соединений в легколетучие производные в результате химических реакций и последующей их отгонке.
Простая отгонка (выпаривание). Это один из самых простых,
доступных и безреактивных методов абсолютного концентрирования. При выпаривании удаляются вещества, которые находятся в форме готовых летучих соединений. Это могут быть макрокомпоненты (отгонка матрицы) и микрокомпоненты; отгонку последних применяют реже. Отгонку матрицы – растворителя используют при определении микроколичеств элементов и органических соединений в различных типах вод, высокочистых кислотах, органических растворителях. Выпаривание матрицы используют при анализе летучих галогенидов (таблица 4.5.1). Выпаривание часто комбинируют с другими методами концентрирования, например, с экстракцией или сорбцией.
Выпаривание можно проводить разными способами, например, нагреванием снизу (с помощью водяной бани) или сверху (под инфракрасной лампой). Выпаривание матрицы может сопровождаться потерями определяемых микрокомпонентов из-за их летучести, механического уноса пробы с газовой фазой и сорбции стенками посуды. Необходимо соблюдать определенные меры предосторожности. Для уменьшения потерь микрокомпонентов, а также облегчения последующего растворения или сбора концентрата к раствору пробы добавляют небольшое количество растворителя с более высокой температурой кипения или графитовый коллектор. При выпаривании под инфракрасной лампой потери обычно меньше.
|
730 |
Таблица 4.5.1 – Отделение элементов испарением |
|
|
|
Испаряемая форма элемента |
Испаряемые элементы |
|
|
Гидриды |
As, Bi, Ge, Pb, Sb, Se, Sn, Te, S, Cl, Br, I |
|
|
Фториды |
B, Si, Ti, Nb, Ta, V, Mo, W |
|
|
Хлориды и оксихлориды |
Al, As, Cr, Ge, Ga, Hg, Sb, Sn, Ta, Ti, V, Mo, Zr, Cd, Zn |
|
|
Бромиды |
As, Hg, Bi, Sb, Se, Sn |
|
|
Иодиды |
As, Sb, Sn, Te |
|
|
Оксиды |
As, Os, Ru, Re, Se, Te, S, C, H |
|
|
Метилборат |
B |
|
|
Необходимость выпаривания раствора с целью уменьшения его объема и увеличения эффективности концентрирования часто возникает при определении органических соединений. В этом случае кроме перечисленных выше потерь определяемых микрокомпонентов возможны потери вследствие разложения веществ или превращения в другие соединения. Если микрокомпоненты при нагревании разлагаются, прибегают к вакуум-отгонке. Для устранения потерь вследствие окисления отгонку проводят в токе инертного газа. Разновидность испарения – сушка под вакуумом в замороженном состоянии (лиофильная сушка). Этот метод имеет определенные преимущества, он позволяет снижать потери легколетучих веществ при анализе, например, вод.
Как уже сказано, распространена отгонка с предварительным химическим превращением, т. е. после перевода макроили микрокомпонента в легколетучее соединение в результате химической реакции. Один из таких методов – сжигание органических и биологических проб (сухая и мокрая минерализация). Этот метод широко используют в элементном органическом анализе. Например, при подводе воздуха или кислорода проба окисляется, и образуются летучие соединения (CO, CO2, N2, SO2, SO3, H2O). Сжигание осуществляют в трубчатых печах различной конструкции. Летучие компоненты улавливают при помощи адсорбционных систем и определяют.
Методы сухой минерализации имеют много недостатков: возможна потеря легколетучих компонентов, а нелетучие могут уноситься газовой фазой в виде аэрозолей. Для предотвращения потерь минерализацию проводят в автоклавах в атмосфере кислорода. Для сухой минерализации можно использовать низкотемпературную кислородную плазму.
При мокрой минерализации потери легколетучих компонентов обычно меньше. Для перевода веществ в раствор применяют концентрированные кислоты и их смеси, различные окислители (H2O2, KClO3, KMnO4) в кислотной и щелочной средах. Для полного и быстрого растворения труднорастворимых веществ используют автоклавы при повышенных значениях температуры и давления.
731
4.5.9 Количественные характеристики концентрирования
Каждый метод концентрирования имеет свои количественные характеристики. При описании любого метода концентрирования используют по крайней мере три величины: степень извлечения, коэффициент концентрирования, коэффициент разделения.
Степень извлечения R – это безразмерная величина, показывающая, какая доля от абсолютного количества микрокомпонента сосредоточена в концентрате:
(4.5.4)
(здесь qK и qпр – абсолютные количества микрокомпонента в концентрате и пробе).
Степень извлечения чаще выражают в процентах:
(4.5.5)
Степень извлечения обычно составляет менее 100% (или менее 1, если не переводят в проценты), поскольку микрокомпонент может теряться на стадиях разложения пробы и концентрирования вследствие испарения или неполного отделения, неполного разложения пробы, неаккуратных действий экспериментатора и значительной сорбции микрокомпонента стенками посуды и аппаратуры.
Как правило, при работе с низкими концентрациями опасность потерь возрастает. В неорганическом анализе в большинстве случаев необходимо достигать степени извлечения микрокомпонентов более чем 95% или, по крайней мере, 90%.
Коэффициент концентрирования, или фактор обогащения К
показывает, во сколько раз изменилось отношение абсолютных количеств микрокомпонента и матрицы в концентрате и в исходной пробе:
(4.5.6)
где Qк и Qпр – абсолютные количества матрицы в концентрате и пробе, qк и qпр – абсолютные количества микрокомпонента там же,
Rмикр и Rматр – степень извлечения микрокомпонента и матрицы.

732
Таким образом, коэффициент концентрирования можно выразить через отношение степеней извлечения микрокомпонента и матрицы. При любом практически полезном концентрировании Rмикр = 1, так что
(4.5.7)
Если степень извлечения равна 1 (R = 100%), формула упрощается:
(4.5.8)
Коэффициент разделения S есть величина, обратная коэффициенту концентрирования:
(4.5.9)
Наряду с разделением компонентов и концентрированием микрокомпонентов важное значение имеет очистка – операция, при которой нужно сохранить основу (макрокомпонент), отбросив примеси.
734
Например, при определении железа его осаждают в виде Fe(OH)3 раствором аммиака. Тригидроксид железа прокаливают и переводят в Fe2O3 и уже по массе оксида железа(III), т.е. Fe2O3, определяют содержание железа. В данном случае осаждаемой формой будет Fe(OH)3, а гравиметрической – оксид железа Fe2O3, т.к. по массе этого осадка рассчитывают содержание железа во взятой навеске. При определении кальция осаждаемой формой является оксалат кальция СаС2О4, а гравиметрической формой – оксид кальция СаО. Осаждаемая и гравиметрическая формы могут совпадать. Барий осаждают в виде BaSO4 и взвешивают также в виде BaSO4, так как при прокаливании его химический состав не изменяется.
В основе гравиметрических определений лежат химические реакции:
-реакции разложения,
-замещения,
-обмена,
-образования комплексных соединений. Разновидности гравиметрического анализа
Пробирный анализ совокупность приемов для определения
драгоценных металлов в сплавах и рудах (наиболее ранний).
Электрогравиметрический анализ – определяемые элементы выделяют из раствора с помощью электролиза, а потом взвешивают.
Термогравиметрия с помощью термовесов позволяет наблюдать, как изменяется масса твердых тел в широком интервале температур (около 1000 °С). Изменение массы пробы при повышении температуры автоматически регистрируется в виде ступенчатой кривой.
(Термогравиметрически показано, что кристаллогидрат оксалата кальция СаС2О4∙Н2О устойчив до температуры 100 °С. При повышении температуры до 226 °С он переходит в безводную соль СаС2О4, при 420 °С оксалат переходит в карбонат кальция СаСО3, при 660 °С карбонат распадается на оксид кальция СаО и диоксид углерода СО2. Процесс заканчивается при 840°С.) Гравиметрическим методом был установлен химический состав большого числа веществ. Он являлся основным методом определения атомных масс. Его используют для определения гигроскопической влаги у широкого круга веществ, кристаллизационной воды, сульфат-иона, диоксида кремния, щелочных, щелочно-земельных и многих других металлов. Метод этот хорошо изучен, но в практике современного анализа применяется сравнительно редко. Его основной недостаток – длительность его проведения.
4.6. 1.2 Типы гравиметрических определений
1. Определяемую составную часть выделяют и взвешивают.
При определении зольности различных материалов на аналитических весах взвешивают небольшой образец топлива навеску (mн). Навеску сжигают в тигле и тщательно прокаливают до тех пор, пока масса золы не прекратит уменьшаться. По точной массе золы (mз) легко вычислить

736
Цель осаждения – количественно перевести определяемую составную часть в малорастворимое соединение – в осаждаемую форму. Осаждаемая и гравиметрическая формы должны соответствовать следующим требованиям.
Требования к осаждаемой форме:
1.Осадок должен быть практически нерастворим: растворимость не должна превышать 1∙10–4–1∙10–5 моль/л, в растворе после осаждения не должно оставаться более 0,1 мг определяемого элемента.
2.Осадок должен быть по возможности крупнокристаллическим. Он должен быть в форме, удобной для отделения его от раствора.
3.Осадок не должен поглощать из раствора различные примеси.
4.Осадок должен иметь постоянный состав.
5.Осаждаемая форма должна легко и полно превращаться в гравиметрическую форму.
Требования к гравиметрической форме:
1.Состав весовой формы должен точно соответствовать её химической формуле (стехиометрический состав).
2.ВФ должна обладать химической устойчивостью к компонентам воздуха (пары воды, кислород, СО2 и т.д.) и продуктам сгорания фильтра.
3.Должна быть термически устойчивой в широком интервале температур.
4.Желательно малое содержание определяемого элемента в весовой форме (по возможности минимальное значение гравиметрического фактора пересчета), чтобы погрешности в определении её массы в меньшей мере сказывались на результатах анализа.
5.Желательно – негигроскопичной.
Выбор осадителя. Осадитель должен быть (желательно) летучим веществом или легко удаляемым. (Поэтому ионы Ва2+ осаждают H2SO4, а не Na2SO4 или K2SO4, а ионы Ag+ осаждают действием НCl, а не NaCl, ионы Fe3+
– действием NH4OH, а не NaOH.) Желательно, чтобы осадитель был специфичен, т. е. осаждал бы данный ион и не осаждал бы другие присутствующие в растворе ионы. (Например, в присутствии ионов Fe3+ ионы А13+ осаждают тиосульфатом натрия Na2S2O3, в отсутствие же ионов Fe3+ ионы А1 осаждают NH4OH, как, соответственно, и ионы Fe3+ в отсутствие ионов А13+.)
Осадок должен иметь как можно меньшую растворимость.
О растворимости осадков можно судить по произведению растворимости для однотипных соединений. Произведение растворимости (ПР) есть произведение концентраций ионов в насыщенном растворе малорастворимого электролита при данной температуре. Например, малорастворимые соли бария имеют следующие значения ПР:
-ВаС2О4 – 1,6∙10–7;
-ВаСО3 – 8,0∙10–9;
-ВаСrО4 – 2,4∙10–10;
-BaSO4 – 1,1∙10–10.
737
Наименьшее значение ПР имеет BaSO4, поэтому определение бария с наименьшими потерями можно вести в виде BaSO4.
Полнота осаждения. Образование осадка происходит тогда, когда произведение концентраций ионов превысит ПР осаждаемого соединения при данной температуре. Нельзя использовать слишком большой избыток осадителя. Это может вызвать повышение растворимости осадка из-за образования кислых солей, комплексных соединений или же проявления амфотерных свойств в случае гидроксидов. Например, прибавление избытка H2SO4 при осаждении PbSO4 может вызвать частичное растворение осадка по реакции PbSO4 + Н2SO4=Pb(HSO4)2. При осаждении ионов Ag+ соляной кислотой в виде AgCl растворимость осадка AgCl повышается вследствие образования комплексного соединения H[AgCl2]. Иногда комплексообразование даже препятствует использованию реакции для целей гравиметрического анализа. Например, осадок HgI2 при малейшем избытке KI переходит в раствор вследствие образования комплексной соли по уравнению
HgI2+2KI = K2[HgI4].
Поэтому эмпирическое правило о полуторном избытке осадителя нельзя применить к данной системе. При осаждении ионов Аl3+ в виде гидроксида избыток реактива может привести к растворению осадка вследствие амфотерности данного гидроксида:
А1(ОН)3 + ОH–=АlO2–+ 2Н2О.
Во многих случаях прибавление небольшого избытка осадителя вызывает значительное понижение растворимости и способствует полноте осаждения.
На полноту осаждения влияют:
-ПР,
-недостаток или избыток осадителя,
-температура (увеличение или снижение растворимости при повышении температуры),
-кислотность или щелочность среды (концентрация ионов Н+),
-солевой эффект (в избытке кислоты),
-возможность комплексообразования (в избытке кислоты),
-способность к коллоидообразованию (коллоидные растворы образуют гидрофильные вещества: А1(ОН)3, Fe(OH)3, AgCl; в таких случаях необходимы меры по разрушению КР: энергичное перемешивание и
нагревание, добавление электролитов, напр. разбавленной HNO3 при осаждении AgCl и т. п.), дисперсности частиц, размеры частиц осадка (крупные кристаллы обладают меньшей растворимостью, чем мелкие).
Механизм образования осадков. В первый момент образуются
738
чрезвычайно мелкие зародышевые кристаллы, которые не могут еще выпасть в осадок. В дальнейшем идет процесс укрупнения зародышевых кристаллов. Он протекает двумя различными путями: в одном случае образуются кристаллические осадки, в другом – аморфные. Если выделение вещества из раствора преимущественно идет на поверхности зародышей кристаллов и последние постепенно растут, то в дальнейшем возникает кристаллический осадок. Если зародышевые кристаллы соединяются в более крупные агрегаты и оседают на дно, то образуется аморфный осадок. Аморфные осадки фактически состоят из мельчайших кристаллов. Особенно легко образуют аморфные осадки малорастворимые вещества. Чем больше концентрация раствора и чем меньше растворимость данного вещества, тем больше возникает зародышевых кристаллов. Если зародышей очень много, то образуются мелкие кристаллы (при быстром смешивании концентрированных растворов). Если растворы будут разбавленными и реактивы смешиваются постепенно, то зародышей образуется меньше, число образовавшихся кристаллов также будет меньше, но они будут крупными. Аморфные кристаллы имеют склонность превращаться в кристаллические. Облик одного и того же кристалла в процессе роста может изменяться. При этом грани передвигаются, но углы между ними сохраняются. Но, достигнув определенных размеров, кристаллы в дальнейшем практически не растут. В случае, когда зародыши кристаллов объединяются в крупные агрегаты, образуются аморфные осадки, которые являются как бы скрыто кристаллическими.
Условия осаждения кристаллических осадков. Для аналитических целей лучше работать с крупнокристаллическими осадками. Они легко отфильтровываются, меньше адсорбируют посторонние вещества. Мелкие кристаллы способны проходить через поры фильтра, что ведет к потере осадка и искажает результаты анализа. Учитывая особенности механизма образования кристаллических осадков, можно создать условия, которые способствуют получению более крупных кристаллов. Более крупные кристаллы образуются в таком растворе, который содержит меньше зародышевых кристаллов. Меньше зародышевых кристаллов будет тогда, когда при осаждении используют разбавленные растворы и при этом осадитель добавляют очень медленно, а в начальной стадии только по каплям. Росту образовавшихся зародышевых кристаллов способствует перемешивание раствора, в местах перемешивания не успевают возникнуть новые зародышевые кристаллы. Для уменьшения степени пересыщения раствора нужно повысить растворимость осадка.
Для этого осаждение проводят при нагревании из горячих растворов. Повышение температуры способствует быстрому растворению мелких кристаллов, и за счет этого увеличению крупных кристаллов. Другой фактор повышения растворимости – понижение рН раствора (увеличение кислотности среды). (Например, при осаждении BaSO4 добавляют HNO3, а осаждение кальция в виде оксалата ведут в кислом растворе.) Для
739
достижения возможно большей полноты осаждения в конце операции повышенную растворимость вновь понижают. Это достигается добавлением избытка осадителя и регулированием кислотности раствора. Несмотря на указанные меры, известная часть осадка выпадает в виде мелких кристаллов, способных проходить через поры фильтра. Но если осадок выдержать несколько часов или еще лучше до следующего дня, то при этом он претерпевает так называемое созревание, в ходе которого мелкие кристаллы растворяются и за счет этого увеличиваются более крупные. Созреванию способствуют повышенная температура и перемешивание раствора. Учет всех факторов позволяет получить осадки, которые хорошо отфильтровываются, и дает возможность избежать потери вещества за счет повышенной растворимости.
Условия осаждения аморфных осадков. Аморфные осадки возникают за счет слипания коллоидных частиц в крупные агрегаты, которые оседают из раствора в виде хлопьев. Поэтому при работе с аморфными осадками важно предотвратить пептизацию и вызвать коагуляцию. Для этого осаждение ведут в присутствии соответствующего электролита-коагулятора (эту роль очень часто выполняют различные соли аммония и кислоты). Коагуляции способствует повышение температуры раствора, т.к. при этом разрушается гидратная оболочка коллоидных частиц и уменьшается адсорбция ионов, которые придают частицам электрический заряд (за счет этого заряда и за счет гидратной оболочки коллоидные частицы удерживаются во взвешенном состоянии и не выпадают в осадок).
Поэтому осаждение аморфных осадков ведут из нагретого анализируемого раствора горячим раствором осадителя. При использовании разбавленных растворов аморфные осадки получаются рыхлыми, с очень большой поверхностью и поэтому сильно адсорбируют посторонние вещества. Чтобы исключить это нежелательное явление, осаждение аморфных осадков ведут из достаточно концентрированных растворов. При этом раствор осадителя прибавляют быстро. Усиление адсорбции за счет увеличения концентрации устраняют прибавлением большого объема горячей воды сразу же после завершения осаждения. При стоянии усиливается процесс адсорбции посторонних примесей большой поверхностью аморфного осадка, ему не дают стоять после осаждения, а сразу же отфильтровывают.
4.6.1.4 Операции гравиметрического анализа
Гравиметрические методы, связанные с получением осадков, включают следующие операции:
1)отбор средней пробы;
2)взятие навески;
3)растворение навески;
4)осаждение определяемой составной части;
5)фильтрование и промывание осадков;
740
6)высушивание и прокаливание осадков;
7)взвешивание осадков;
8)вычисление результатов анализа.
Отбор средней пробы. Состав отобранной средней пробы должен приближаться к среднему химическому составу большого количества исследуемого материала.
Взятие навески. Из отобранной для анализа средней пробы, отражающей состав исследуемого материала, или же из предварительно очищенного от примесей вещества берут навеску. Навеска представляет собой строго определенное количество вещества, необходимое для выполнения анализа.
При выборе размера навески учитывают:
1)метод, с помощью которого проводят определение (грамм-метод, сантиграмм-метод, миллиграмм-метод);
2)при большой навеске достигается более высокая относительная точность определения;
3)при больших навесках осадок трудно отфильтровывать, промывать, прокалить;
4)при большой навеске удлиняется время выполнения анализа;
5)при малых навесках снижается точность определения.
При определении размера навески исходят из количества осаждаемой формы. При обычных гравиметрических определениях масса аморфных осадков должна быть около 0,1 г, легких кристаллических осадков – около 0,1-0,2, тяжелых кристаллических осадков – 0,2-0,4, очень тяжелых кристаллических осадков – около 0,4-0,5 г. При определении влажности или зольности различных материалов берут навеску в 1,0 2,0 г и даже больше. При определении содержания примесей порядка 0,001% навеску увеличивают до нескольких граммов и даже до нескольких десятков граммов. Вещества, которые не выделяют паров и не поглощают из воздуха его составных частей, взвешивают на часовом стекле. Вещества, способные выделять пары и взаимодействующие с атмосферой, взвешивают в бюксах.
Растворение навески. Навеску переносят в чистый химический стакан или коническую колбу. В качестве растворителей используют дистиллированную воду, кислоты, смеси кислот, щелочи. Количество кислоты или щелочи, необходимое для растворения навески, рассчитывают по уравнению реакции с учетом концентрации растворителя. При растворении может энергично выделяться газ. Пузырьки газа могут уносить капельки раствора. Чтобы исключить потери, стакан накрывают часовым стеклом, а в отверстие конической колбы вставляют воронку с короткой шейкой.
Выпаривание (уменьшение объема раствора) проводят химическом стакане или фарфоровой чашке на водяной бане (температура не должна превышать 100 °С) или на песочной бане.
Осаждение. Осаждение проводят в стаканах вместимостью 200-250 мл.
741
В большинстве случаев его ведут из горячих растворов. Необходимое количество осадителя берут в соответствии с расчетом. Для полноты реакции добавляют избыток растворителя 50% для нелетучих растворителей и 100200% для летучих растворителей. Для медленного добавления осадителя его наливают в бюретку со стеклянным краном, соответственно регулируя скорость вытекания. После осаждения и просветления жидкости над осадком проверяют полноту осаждения. Для этого 2-3 капли раствора осадителя прибавляют по стенке стакана и наблюдают появление мути в месте смешивания. При появлении даже легкой мути добавляют несколько миллилитров осадителя, перемешивают раствор стеклянной палочкой и снованагревают. После просветления жидкости вновь проверяют полноту осаждения. Кристаллические осадки следует фильтровать через несколько часов после осаждения, а еще лучше на следующие сутки. Аморфные осадки отфильтровывают горячими через 10-15 мин после осаждения. К фильтрованию приступают тогда, когда жидкость над осадком становится совершенно прозрачной.
Фильтрование и промывание осадков. Аморфные и кристаллические осадки отфильтровывают через беззольные бумажные фильтры. Через бумажные фильтры не рекомендуется отфильтровывать осадки, которые разлагаются при сжигании (например, AgCl).
По плотности бумаги различают 3 сорта бумажных фильтров:
1)черная или красная лента – наименее плотные для отделения аморфных осадков гидроксидов, таких, как А1(ОН)3, Fe(OH)3 и др.;
2)белая лента – средней плотности для отделения большинства кристаллических осадков;
3)синяя лента – наиболее плотные для отделения мелкокристаллических осадков (BaSO4, CaC2O4 и др.).
Сначала на бумажный фильтр по палочке выливают из стакана маточный раствор. Оставшийся в стакане осадок взмучивают, добавляя небольшие порции дистиллированной воды или промывной жидкости. Эту операцию повторяют 2-3 раза. Затем осадок переносят на фильтр. Для этого его смешивают с небольшим количеством дистиллированной воды или промывной жидкости. Образующуюся суспензию переливают по стеклянной палочке на фильтр. Для удаления частиц осадка, приставших к стенкам стакана, их протирают стеклянной палочкой с чистым каучуковым наконечником или маленькими кусочками беззольного фильтра. Эти кусочки потом бросают в фильтр с осадком. В стакане и на стеклянной палочке не должно быть частиц осадка. После перенесения всего осадка на фильтр приступают к его промыванию на фильтре. Для этого струю жидкости из промывалки направляют в воронку. Когда фильтр заполняется наполовину, жидкости дают стечь полностью с фильтра. Эту операцию повторяют несколько раз. При этом струю из промывалки направляют по краям фильтра сверху вниз по спирали, пока осадок не будет собран в глубине фильтра (рисунок 4.6.1).

742
Рисунок 4.6.1 – Промывание
Промывание заканчивают, когда проверочная реакция покажет отрицательный результат на присутствие ионов-примесей. Стеклянные фильтрующие тигли удобны для отделения кристаллических осадков, если их не прокаливают, а высушивают. Для ускорения работы фильтрование можно вести под вакуумом. Через фильтрующие тигли нельзя отфильтровывать студенистые осадки.
Высушивание, прокаливание и взвешивание осадков. Воронку с осадком закрывают фильтровальной бумагой и помещают в сушильный шкаф. При этом полное высушивание осадка не обязательно и не желательно, так как при складывании сухих фильтров с осадком возможны потери осадка в виде мелкой пыли. Слегка влажный фильтр легче укладывать в фарфоровый тигель. Фильтр с осадком осторожно переносят в тигель пинцетом с полиэтиленовым наконечником или свертывают в спираль (рисунок 4.6.2) и помещают в подготовленный тигель. Фарфоровый тигель должен быть предварительно доведен до постоянной массы.
Примечание: а, б, в – сгибание краев фильтра; г – свертывание в спираль; д – свернутый фильтр с осадком
Рисунок 4.6.2 – Свертывание фильтра с осадком
Для прокаливания осадков применяют только фарфоровые фильтрующие тигли (выдерживают температуры от 300 до 1000 °С). Прокаливание ведут на газовой горелке или в муфельной печи. После прокаливания раскаленный тигель переносят щипцами на гранитную плиту приблизительно на 30 с. Затем тигель помещают в эксикатор приблизительно на 30 мин (до полного охлаждения). В эксикаторе тигель переносят в

743
весовую комнату и взвешивают. После взвешивания прокаливают еще 15-20 мин, затем вновь охлаждают в эксикаторе и опять взвешивают. Так продолжают до получения постоянной массы. Постоянная масса считается достигнутой тогда, когда разность между предыдущей и последующей массой составляет 0,0001-0,0002 г. Наименьшее из двух таких чисел берут как окончательное.
4.6.1.5 Расчеты в гравиметрическом анализе
В гравиметрическом анализе рассчитывают:
1)размер навески;
2)количество растворителя, необходимое для растворения навески;
3)количество осаждаемого реактива;
4)результаты анализа.
Расчеты по 1, 2 и 3-му пунктам ведут приближенно. В этом случае необходимо знать 1-2 значащие цифры. Вычисление результатов анализа (п. 4) ведут с той точностью, которая отвечает точности взвешивания.
Пример 1. Рассчитать размер навески железной руды, содержащей около 25% железа. Железо будет осаждаться в виде Fe(OH)3.
Поскольку осадок Fe(OH)3 является аморфным, его масса должна быть около 0,1 г. Вычислим, сколько железа необходимо, чтобы иметь 0,1 г осадка Fe(OH)3. Для этого составим соответствующую пропорцию, беря округленные значения молярной массы Fe(OH)3 (107 г/моль) и молярные массы Fe (56 г/моль):
Найдем теперь, сколько нужно взять руды, чтобы в ней было 0,05 г Fe. Для этого составим новую пропорцию, в которой через у обозначим
размер навески:
Размер навески должен быть около 0,2 г. Расчет количества растворителя для растворения навески и количества осаждающего реактива также ведут, опираясь на закон эквивалентов и на использование пропорций. Для обеспечения полноты реакции нелетучих реактивов берут в полтора раза больше, а летучих – в 2-3 раза больше.
Пример 2. Какой объем 2 М раствора аммиака необходим при тройном избытке для осаждения ионов Fe из раствора, если было растворено 0,1 г железа?
Напишем уравнение реакции:

744
Из равенства видно, что на 56 г железа необходимо 105 г NH4OH. Определим, в каком объеме содержится 105 г NH4OH, исходя из
формулы m=cMV.
После подстановки имеем 105 = 2-35 V. Отсюда К=1,5л, или 1500 мл. Объем, необходимый для осаждения 0,1 г железа в соответствии с уравнением реакции, найдем из пропорции:
При тройном избытке необходимо взять 2,7∙3 ≈ 8 мл 2 М раствора
NH4OH.
Результаты гравиметрических определений обычно выражают в процентах, реже – в единицах массы. Если анализируют металлы или их сплавы, то результат относят к химическим элементам, например, %Fe, %Mn, %С, %S. Если анализируют силикаты, горные породы и другие вещества, содержащие кислород, то результат анализа выражают в виде содержащихся в них оксидов, например, %SiO2, %А12О3, %Fe2O3, %FeO, %CaO, %P2O5,
%К2О и т.д. Если определяемая составная часть (элемент, вода, зола, оксид) выделена в той форме, в какой выражают ее содержание в пробе, то для нахождения содержания х (в %) используют формулу
(4.6.4)
где m0 – масса выделенной составной части; mн – масса навески.
4.6.2 Титриметрические методы анализа 4.6.2.1 Характеристика титриметрического анализа
Титриметрический анализ – метод количественного анализа, в котором измеряют количество реактива, затраченного в ходе химической реакции.
Втитриметрическом анализе используют точное измерение объемов реагирующих веществ.
Титриметрические методы дают большой выигрыш во времени, поэтому их так широко используют в химических лабораториях.
Втитриметрическом анализе реакцию проводят между двумя растворами и как можно точнее определяют момент завершения реакции между обоими веществами. Зная концентрацию одного, можно установить и точную концентрацию другого.
Гравиметрический и титриметрический анализы основаны на законе эквивалентов.
(4.6.5)
746
2.Методы окисления-восстановления, основанные на взаимодействии между окислителем и восстановителем.
3.Методы комплексообразования, основанные на образовании малодиссоциирующих комплексных ионов или молекул.
4.Методы осаждения, основанные на реакциях образования малорастворимых соединений.
5.Методы, основанные на использовании реакций с применением
ионитов.
В зависимости от способа определения (или индикации) конца титрования выделяют:
1.Индикаторное титрование, основанное на применении индикаторов, т. е. веществ, способных менять окраску в конце титрования.
2.Потенциометрическое титрование, в котором роль индикатора выполняет электрод, потенциал которого зависит от концентрации одного из веществ, взаимодействующих в данной реакции.
3.Амперометрическое титрование. В этом случае раствор помещают в электролизер, снабженный капельным ртутным катодом и большим ртутным анодом. При титровании уменьшается как концентрация свободных ионов металла, так и сила тока. Наиболее резкий скачок наблюдается в конце титрования. Метод применяют для определения катионов, анионов и органических веществ. Кроме капельного ртутного электрода применяют твёрдые микроэлектроды.
4.Кондуктометрическое титрование, основанное на изменении электрической проводимости растворов в процессе титрования. Сначала электрическая проводимость может падать, а затем вновь возрастать. Для расчета строят график, на котором пересекаются две прямые; точка пересечения соответствует эквивалентности.
5.Высокочастотное титрование, в котором для установления конечной точки титрования используют переменные токи высокой частоты. Электроды не соприкасаются с раствором, поэтому Данный метод называют еще безэлектродной кондуктометрией.
6.Оптические методы, основанные на измерении светопоглощения в процессе титрования. Они применимы в тех случаях, когда существует линейная зависимость между светопоглощением и концентрацией определяемого вещества в анализируемом растворе.
В зависимости от приемов определения выделяют:
1.Прямое титрование, когда непосредственно (прямо) реагируют два раствора, концентрация одного из которых известна, а концентрация другого неизвестна. Например, раствор кислоты (НС1, H2SO4) с известной концентрацией приливают по каплям к раствору щелочи с неизвестной концентрацией. В большинстве случаев используют прямое титрование.
2.Обратное титрование (титрование по остатку). В некоторых случаях нельзя применить прием прямого титрования, например, если раствор А не реагирует с раствором В или же реагирует очень медленно, но существует
747
третье вещество С, которое реагирует с А и В. В этом случае осуществляют реакцию между веществом А и веществом С, которое берут с заведомым избытком, и избыток вещества С заставляют прореагировать с веществом В. Если концентрации веществ В и С известны, то можно определить и концентрацию вещества А.
3. Метод замещения. Если вещества А и В не взаимодействуют между собой, можно найти вещество С, которое при взаимодействии с веществом А выделяют эквивалентное количество вещества D, которое взаимодействует с веществом В. По количеству выделенного вещества D можно определить количество вещества А.
4.6.2.4 Кислотно-основное титрование 4.6.2.4.1 Теоретические основы метода
В основе метода лежит реакция нейтрализации: Н+ + ОН-Н2О
Метод применяется для количественного определения кислот и щелочей, а также для проведения титриметрических определений, связанных с методом нейтрализации, например, определение некоторых солей, образованных сильными основаниями и слабыми кислотами (Na2CO3, Na2B4O7) или солей аммония.
При количественном определении кислот алкалиметрии – рабочим раствором является раствор щелочи NaOH или KOH. Приготовить титрованный раствор щелочи по навеске невозможно, кроме того, даже при самом тщательном хранении раствора щелочей легко и быстро меняют свой титр, поэтому титр этих рабочих растворов устанавливают. Исходными веществами для установки титра рабочего раствора щелочи может служить щавелевая кислота (H2C2O4 × 2H2O) или янтарная кислота (H2C4H4O4). Часто используют децинормальные растворы кислоты (0,1 Н), приготовленные из фиксанала.
При количественном определении щелочи ацидиметрии – рабочим раствором является раствор сильной кислоты (HCl, H2SO4). Приготовить титрованный раствор кислоты, исходя из концентрированной, невозможно, поэтому титр устанавливают.
Исходным веществом для установки титра кислоты служит бура (Na2B4O7×10H2O) или сода (Na2CO3), В некоторых случаях рабочий раствор готовят из фиксанала.
Кислотно-основной метод используют для определения кислотности желудочного сока, для определения карбонатной жесткости воды, кислотности молочных продуктов, квашенной капусты, безалкогольных напитков. В фармацевтическом анализе – для определения концентрации HCl, количества гидрокарбоната, борной кислоты и др.
Если титровать растворы кислоты раствором щелочи, происходит связывание ионов Н+ ионами ОН- и концентрация ионов Н+ постепенно
748
уменьшается, а рН раствора увеличивается. При определенном значении рН достигается точка эквивалентности и титрование должно быть закончено.
При титровании раствора щелочи раствором кислоты связываются ионы ОН- и концентрация их в растворе уменьшается, а концентрация ионов Н+ увеличивается и рН раствора уменьшается. Однако значение рН в точке эквивалентности не во всех случаях имеет одно и то же значение – оно зависит от природы реагирующих кислоты и основания.
При нейтрализации сильной кислоты сильным основанием образуется один слабый электролит – Н2О.
HCl + NaOH NaCl + H2O
Реакция практически доходит до конца. NaCl гидролизу не подвергается и имеет нейтральную реакцию, т.е. при титровании сильной кислоты щелочью, и наоборот, в точке эквивалентности среда раствора нейтральная, рН = 7.
Если вместо сильной кислоты титровать щелочью слабую кислоту (уксусную), то происходит реакция:
CH3COOH + NaOH CH3CONa + H2O
В растворе в точке эквивалентности будет присутствовать соль, подвергающаяся гидролизу:
CH3COONa + H2O CH3COOH + NaOH
Следовательно, реакция обратима и не будет доходить до конца. Титрование будет закончено при рН > 7.
При титровании соабых оснований сильными килотами:
NH4OH + HCl NH4Cl + H2O
Образующаяся соль подвергается гидролизу. Реакция нейтрализации будет обратима и в точке эквивалентности концентрация ионов Н+ превысит концентрацию ионов ОН-. Титрование будет закончено при рН < 7.
Таким образом, в методе нейтрализации лишь при взаимодействии сильной кислоты с сильным основанием точка эквивалентности будет совпадать с точкой нейтрализации. При титровании необходимо устанавливать точку эквивалентности, а не нейтрализации. Следовательно, титрование в разных случаях приходится заканчивать при различных рН.
4.6.2.4.2 Кислотно-основные индикаторы
Точка эквивалентности при реакциях нейтрализации не сопровождается каким-либо внешними изменениями. Поэтому для определения конца реакции применяются специальные индикаторы. В точке

749
эквивалентности происходит изменение рН раствора, поэтому индикаторы, применяемые при кислотно-основном титровании, представляют собой органические соединения, окраска которых меняется в зависимости от концентрации ионов Н+ в растворе. Это кислотно-основные (или рН) индикаторы.
Область значений рН раствора, в которой происходит заметное изменение окраски индикатора, называется областью перехода индикатора. Это наиболее важная характеристика индикатора при нейтрализации – показатель титрования (рТ) – та концентрация ионов Н+, при которой наиболее резко изменяется окраска индикатора (таблица 4.6.1).
Таблица 4.6.1 – Интервал перехода некоторых индикаторов
Пригодность того или иного индикатора для данного титрования можно количественно охарактеризовать индикаторной ошибкой титрования, возникающей вследствие несовпадения рТ и рН в точке эквивалентности. Индикатор изменяет окраску при pH = pT, а точка эквивалентности может не совпадать с этим значением. В этом случае может остаться недоттитрованная кислота или основание (как слабые, так и сильные). Или при перетитровании в растворе могут появляться избыточные протоны H+ или OH– - ионы. Разделяют:
750
Первые две – это ошибки, визникающие после титрования сильных кислот и оснований, две последние – для слабых кислот и оснований.
Выбор индикатора. Чтобы правильно выбрать индикатор, нужно знать, как изменится рН в процессе титрования и каково его значение в точке эквивалентности. В каждом случае титрования следует применять такой индикатор, численное значение показателя титрования которого отличается от рН титруемого раствора в момент эквивалентности.
1.Титрование сильной кислоты слабым основанием или слабого основания сильной кислотой.
Вследствие гидролиза соли в точке эквивалентности накапливается катион Н+, поэтому среда кислая. Титрование нужно закончить в кислой среде. В качестве индикатора можно применить лакмус, метилоранж или метиловый красный, т.к. они меняют окраску при рН < 7.
2.Титрование слабой кислоты слабым основанием или сильного основания слабой кислотой.
В точке эквивалентности накапливаются ОН- ионы, поэтому среда раствора щелочная. Титрование заканчивается в щелочной среде. В качестве индикатора можно применить фенолфталеин, т.к. он меняет окраску в щелочной среде при рН =8,2–10,0
3.Титрование сильной кислоты сильным основанием или сильного основания сильной кислотой.
В момент эквивалентности среда нейтральная, рН = 7. Следовательно, нужно применить индикатор, меняющий окраску при рН = 7. Можно применять индикаторы, изменяющие окраску от 4 до 10 (все индикаторы).
Изменение рН, происходящее по мере нейтрализации различных по степени диссоциации кислот и оснований, обычно изображают графически. Такие графики постепенного изменения рН при нейтрализации называют кривыми нейтрализации или кривыми титрования метода нейтрализации.
4.6.2.4.3 Построение кривых кислотно-основного титрования
Кривая титрования – это графическое изображение зависимости какого-либо параметра системы, связанного с концентрацией определяемого иона, от объема, добавленного титранта. В кислотно-основном титровании этим параметром является рН, т.е. кривая титрования – это зависимость рН титруемого раствора от объема титранта.
Выделяют:
А) область до начала титрования; Б) область до начала скачка титрования;
В) скачок титрования, включая точку эквивалентности; Г) область после скачка титрования.
До начала титрования значение рН титруемого раствора определяется концентрацией и константой диссоциации анализируемой кислоты (или основания)
После точки эквивалентности – концентрацией титранта.
751
В промежуточных точках титрования факторы, определяющие рН, зависят от того какое вещество титруют.
Рассмотрим титрование сильной кислоты сильным основанием
(рисунок 4.6.1). Точка эквивалентности на кривой титрования совпадает с точкой нейтральности. Кривая симметрична относительно точки эквивалентности.
Резкое изменение pH в области точки эквивалентности называют скачком титрования.
Границы скачка устанавливаются в зависимости от требуемой точности титрования. Так, погрешность многих титриметрических методов не превышает ±0,1%. Поэтому если задать точность титрования 0,1%, то скачок титрования – это изменение рН от состояния, когда раствор не дотитрован на 0,1% к состоянию, когда он на 0,1% перетитрован. В этом случае скачок титрования составляет 6 единиц рН (от рН 4 до рН 10).
Рисунок 4.6.1 – Кривая титрования 100 мл 0,1 М HCl 0,1 М раствором NaOH
Если допустимая погрешность анализа составляет ±1%, то скачком титрования можно считать изменение рН от недотитрованого на 1% раствора до перетитрованого на эту величину (в этом случае скачок длится от рН 3 до рН 11, т.е. составляет 7 единиц рН). Расчеты рН показывают, что величина скачка титрования зависит от концентрации реагирующих веществ. Чем меньше концентрация реагентов, тем меньше скачок титрования. Повышение температуры вызывает увеличение ионного произведения воды, и это тоже уменьшает скачок титрования. Причем ветвь кривой титрования до точки эквивалентности при всех температурах остается практически без изменений, в то время как ветвь после точки эквивалентности пойдет ниже.
Кривая титрования сильного основания сильной кислотой
представляет собой зеркальное изображение кривой титрования сильной кислоты сильным основанием.
Далее рассмотрим титрование слабой кислоты сильным основанием

752
(рисунок 4.6.2).
Рисунок 4.6.2 – Кривая титрования 100 мл 0,1 М CH3COOH 0,1 М раствором NaOH
Скачок титрования длится от рН = 7,76 до рН = 10. Точка эквивалентности – при рН = 8,88. Т.е. точка эквивалентности не совпадает с точкой нейтральности. Скачок титрования намного меньше, чем для сильной кислоты, он составляет всего 2,3 единицы рН вместо 6.
Величина скачка титрования на кривых титрования зависит от:
1.Концентрации титруемого раствора. Чем она меньше, тем меньше скачок титрования. Именно поэтому практически невозможно оттитровать раствор сильных кислот слабее чем 10–4 М и растворы слабых кислот слабее чем 10–2 М.
2.Температура. С изменением температуры изменяется величина Kw.
Врезультате этого с ростом температуры скачок титрования уменьшается.
3.Природа титруемой кислоты (или основания).
С уменьшением константы диссоциации кислоты скачок титрования уменьшается. Практически невозможно зафиксировать скачок титрования меньший, чем 2 единицы рН (это обусловлено величиной интервала перехода индикатора). Следовательно, нельзя оттитровать растворы кислот, имеющие Kк<10–9. Это значит невозможно получить отчетливый скачок, например, при титровании борной кислоты H3BO3 (K = 5,6·10-10).
4.6.2.5 Окислительно-восстановительное титрование
Методы окислительно-восстановительного титрования (методы редоксиметрии) широко используются в аналитической химии, а также в анализе лекарственных препаратов.
Окислительно-восстановительное титрование основано на реакциях окисления-восстановления.
Для количественного анализа подходят те реакции, которые отвечают следующим требованиям:
753
1.Протекают до конца;
2.Проходят быстро;
3.Образуют продукты определенного (известного) состава;
4.Позволяют фиксировать точку эквивалентности;
5.Не дают побочных реакций.
В количественном анализе используются реакции у которых константа равновесия равна или больше 108 (КР>108). Методы окислительновосстановительного титрования называют по типу применяемого титранта. Например, если титрантом является перманганат калия KMnO4 – метод называется перманганатометрия; если титрантом является нитрит натрия NaNO2 –метод называется нитритометрия; если титрантом является бромат калия KBrO3 – метод называется броматометрия и т.д.
4.6.2.5.1 Перманганатометрия
Метод перманганатометрии основан на реакциях окисления восстановителей перманганат-ионом MnO4- . Окисление может проводиться
вкислой среде, или в нейтральной или слабо щелочной среде.
Вкислой среде восстановление MnO4- до Mn2+ протекает с присоединением 5 электронов.
MnO4-+8 H++5е- Mn2++4H2O
φo(MnO4-/ Mn2+) = + 1,51 B
В нейтральной или слабо щелочной среде перманганат-ионы восстанавливаются до оксида марганца MnO2 , присоединяя 3 электрона.
MnO4-+2H2O + 3е- MnO2 + 4OH-
φo(MnO4-/ MnO2) = + 0,59 B
Вкислой среде молярная масса эквивалента перманганата калия равна:
M (1/z, KMnO4) = 1/5 M (KMnO4) = 1/5 * 158,034 = 31,6068 ~ 31,61
г/моль В нейтральной и слабо щелочной среде молярная масса эквивалента
перманганата калия равна:
M(1/z, KMnO4) = 1/3 M (KMnO4) = 1/3 * 158,034 = 52,678 52,68 г/моль Стандартный потенциал пары MnO4-/ Mn2+ гораздо выше, чем стандартный потенциал пары MnO4-/ MnO2. Следовательно, в кислой среде
перманганат калия является более сильным окислителем.
Раствор перманганата калия фиолетового цвета. При титровании в кислой среде образуются бесцветные катионы марганца Mn2+. Если титрование проводить в нейтральной или слабо щелочной среде, то образуется коричневый осадок оксида марганца MnO2, который затрудняет фиксирование точки эквивалентности. Поэтому в аналитической химии чаще используют перманганатометрическое титрование в кислой среде.
754
Титрованным раствором в методе перманганатометрии является раствор перманганата калия KMnO4. Кристаллический перманганат калия всегда содержит в качестве примесей некоторое количество MnO2 и другие продукты разложения. Кроме того, перманганат калия легко вступает в редокс-реакции с органическими веществами, которые попадают в дистиллированную воду.
Концентрация раствора перманганата калия в первое время после приготовления немного уменьшается. Поэтому, приготовленный раствор перманганата калия, оставляют в темном месте на 7–10 дней. За это время происходит окисление восстановителей, присутствие которых в дистиллированной воде полностью исключить не удается (пыль, следы органических соединений и др.). Затем раствор фильтруют и стандартизируют.
Эталонными веществами для перманганата калия являются: дигидрат щавелевой кислоты H2C2O4•2H2O, оксалат натрия Na2C2O4, оксалат аммония (NH4)2C2O4•H2O, металлическое железо Fe, гексацианоферрат (II) калия
K4[Fe(CN)6].
Например, приготовили 500,0 мл раствора перманганата калия KMnO4 с молярной концентрацией эквивалента С(1/z) 0,1 моль/л. Нужно установить его точную концентрацию. Для этого готовят раствор эталонного вещества, например, дигидрата щавелевой кислоты. Берут точный объем (10,00 мл) эталонного раствора, добавляют серную кислоту для создания кислой среды и титруют приготовленным раствором KMnO4.
5Н2С2О4 + 2KMnO4 + 3 H2SO4 10CO2 + 2MnSO4 + K2SO4 + 8Н2О
Но так как реакция между перманганатом калия и щавелевой кислотой имеет маленькую скорость (проходит очень медленно), то эталонный раствор нагревают до 70-80оС. Горячий раствор медленно, по каплям начинают титровать перманганатом калия. Первые капли перманганата калия даже в горячем растворе обесцвечиваются очень медленно. Но в ходе титрования концентрация ионов Mn2+ возрастает и скорость реакции увеличивается.
Реакция сама делает катализатор. Такие реакции называются автокаталитическими. Если прибавлять большое количество титранта, то может проходить побочная реакция, в результате которой образуется коричневый осадок MnO2.
Хранить титрованный раствор перманганата калия нужно в темном месте или в склянке темного стекла, так как свет ускоряет разложение
KMnO4.
4KMnO4 + 2H2O 4MnO2 + 3O2 + 4KOH
Индикатором в перманганатометрии является сам титрованный раствор перманганата калия, который имеет фиолетовый цвет. При малейшем
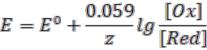
755
избытке KMnO4 раствор окрашивается в розовый цвет. Поэтому в методе перманганатометрии используют безиндикаторное титрование. Титрование заканчивают, когда одна капля титранта вызывает появление бледнорозового окрашивания раствора, не исчезающего в течение 30 секунд.
Так как титрование проводят в кислой среде, то обязательно нужно добавлять при титровании кислоту – серную H2SO4 или фосфорную H3PO4 . В тех случаях, когда в растворе присутствуют ионы, которые образуют осадки с сульфат-ионами, применяют азотную кислоту. Хлороводородную кислоту HCl использовать нельзя, так как она может вступать в окислительновосстановительную реакцию с KMnO4. Реакция сильно ускоряется в присутствии солей железа.
2KMnO4 + 16 HCl 5Cl2 + 2MnCl2 + 8H2O + 2KCl
В аналитической химии методом перманганатометрии определяют:
-прямым титрованием – восстановители: соли железа (П), щавелевую кислоту и её соли, альдегиды, пероксид водорода и другие вещества.
-обратным титрованием – окислители: железо (Ш), нитраты, бихроматы.
-заместительным титрованием можно определять соли кальция, бария, свинца.
4.6.2.5.2 Кривые окислительно-восстановительного титрования
Процессы окислительно-восстановительного титрования можно представить графически в виде кривых титрования, изображающих изменение окислительно-восстановительного потенциала титруемого раствора по мере приливания к нему стандартного раствора окислителя или восстановителя (титранта). При титровании изменяется соотношение окисленной и восстановленной форм определяемого вещества и титранта, поэтому потенциалы редокс-пар рассчитывают по уравнению Нернста:
(4.6.6)
Расчет ведут по потенциалу той окислительно-восстановительной пары, компоненты которой находятся в избытке в данный момент титрования и концентрация которых легко может быть вычислена.
До точки эквивалентности потенциал рассчитывают по потенциалу пары определяемого вещества, а после точки эквивалентности – по системе титранта.
В точке эквивалентности потенциал системы определяется присутствием окислительно-восстановительных пар как определяемого вещества, так и титранта.
Поэтому потенциал в точке эквивалентности, Еэкв, можно рассчитать

756
по уравнению, полученному суммированием уравнений потенциалов обеих пар при условии равенства в точке эквивалентности концентраций окисленных и восстановленных форм окислителя и восстановителя:
(4.6.7)
Расчет кривых титрования проводят для нахождения скачка титрования с целью выбора подходящего редокс-индикатора. Правильно выбранным индикатором является тот индикатор, у которого потенциал перехода окраски находится в пределах скачка титрования.
Скачок титрования в окислительно-восстановительном титровании – это резкое изменение потенциала окислительно-восстановительной системы в пределах допустимой погрешности измерения определяемого вещества.
Общий вид кривой титрования представлен на рисунке 4.6.3.
В начале титрования кривая изменяется плавно, а вблизи точки эквивалентности наблюдается резкое изменение потенциала. По резкому скачку кривой титрования устанавливают точку эквивалентности, которая не всегда лежит на середине скачка. Характер кривых титрования в окислительно-восстановительном титровании не зависит от разбавления раствора, если стехиометрические коэффициенты у окислителя и восстановителя одинаковы. Величина скачка титрования зависит от разности стандартных окислительно-восстановительных потенциалов редокс-пар, участвующих в реакции.
Рисунок 4.6.3 – Общий вид кривой окислительно-восстановительного титрования
Область скачка на кривой окислительно-восстановительного титрования можно значительно расширить, регулируя кислотность среды, используя процессы комплексообразования или образования малорастворимых соединений, т. к. в этих случаях изменяется концентрация окисленной или восстановленной форм соответствующих редокс-пар.

757
Для обнаружения конечной точки титрования используют:
-исчезновение или появление окраски титранта или титруемого вещества,
-окислительно-восстановительные и специфические индикаторы,
-инструментальные методы (потенциометрическое титрование и др.) Окислительно-восстановительные индикаторы (редокс-индикаторы)
представляют собой органические соединения, окисленная и восстановленная формы которых имеют различные окраски. Эти индикаторы имеют определенную окраску в пределах определенных значений окислительно-восстановительного потенциала. Интервал перехода редоксиндикаторов (pT) описывается уравнением:
(4.6.8)
Окислительно-восстановительны индикаторы изменяют свою окраску в связи с достижением титруемым раствором определенного значения окислительно-восстановительного потенциала.
Чтобы окраска редокс-индикатора изменялась при титровании резко, и индикаторная погрешность титрования была незначительной, интервал перехода индикатора должен находиться в пределах скачка потенциалов на кривой титрования.
В качестве окислительно-восстановительных индикаторов применяются дифениламин, N-фенилантраниловая кислота, ферроин, метиловый синий и др. Все эти индикаторы являются обратимыми, т. е. при избытке окислителя окрашиваются, а при избытке восстановителя обесцвечиваются и наоборот. При окислении или восстановлении молекула индикатора не разрушается, а лишь меняет строение. Кроме того, существуют окислительно-восстановительные индикаторы, которые разрушаются необратимо при определенном потенциале (например, нейтральный красный, применяемый для броматометрического определения сурьмы).
4.6.2.6 Осадительное титрование
Методы осадительного титрования основаны на взаимодействии определяемого вещества с титрованным раствором осадителя, в результате которого выпадает осадок.
Вметоде осадительного титрования применяются титранты, образующие осадки с определяемыми веществами. При этом используют реакции, дающие осадки с произведением растворимости меньше 10-10, образовавшиеся осадки не выделяют и не взвешивают.
Наиболее широкое применение получили следующие методы осадительного титрования:
1. Аргентометрическое титрование, титрант – раствор АgNО3.
Воснове метода лежит реакция образования трудно растворимого
758
галогенида серебра.
Для определения анионов применяют нитрат серебра, для определения катионов серебра – хлорид натрия. Аргентометрическим методом титрования пользуются главным образом для количественного определения галогенидионов и ионов серебра.
2.Тиоцианометрическое титрование (роданометрическое), титрант– раствор NН4CNS.
Роданометрическим методом титрования пользуются для определения галогенид-ионов и ионов серебра. Для определения катионов Аg+ в качестве стандартного раствора используют роданид (тиоцианат) аммония, для определения галогенидов и других анионов – нитрат серебра и роданид аммония. В качестве индикатора применяют насыщенный раствор железоаммонийных квасцов.
3.Меркурометрическое титрование, титрант – раствор Нg2(NО3)2.
Этот метод основан на реакции осаждения раствором Нg2(NО3)2
различных анионов.
4. Сульфатометрическое титрование, титрант – раствор ВаСl2 или раствор Н2SО4.
Этот метод применяют для анализа солей бария, титруя их раствором Н2SО4 или для определения сульфатов титрованием раствором соли бария.
Точку эквивалентности в методах осадительного титрования определяют химическим путем (индикаторами на избыток титранта или исчезновение определяемого вещества), а также инструментальными методами.
В методе осаждения применяют следующие типы индикаторов:
осадительные, металлохромные (комплексообразующие) и адсорбционные.
Процесс титрования может быть охарактеризован кривой титрования, построенной в координатах: рМ или рГ – V, где рМ или рГ – взятый с обратным знаком логарифм концентрации ионов металла Мn+ или галогена Г- ; V – объем титранта (рисунок 4.6.4). Скачок титрования на кривой титрования зависит от растворимости осаждаемого соединения. Чем меньше растворимость осаждаемого соединения, тем больше скачок титрования.

759
Рисунок 4.6.4 –Кривая осадительного титрования
Осадительное титрование имеет ряд ограничений в использовании. Использование этого метода для количественного определения
анализируемого вещества возможно только в том случае, если выделяющиеся осадки практически нерастворимы, если реакция образования осадка протекает быстро, и если результаты титрования, не искажаются побочными реакциями соосаждения. Реакции осаждения при использовании в титриметрическом методе анализе обладают рядом недостатков:
-процесс осаждения обратим;
-небольшая скорость многих реакций осаждения;
-при образовании осадка определяемого вещества происходят процессы соосаждения, адсорбции, образования коллоидов.
Однако существуют индикаторы, позволяющие фиксировать точку эквивалентности.
4.6.2.7 Комплексометрическое титрование
Комплексометрическое титрование основано на реакциях образования комплексов. Среди реакций с участием неорганических лигандов применяются реакции образования галогенидов ртути (II), фторидов алюминия, циркония, тория и цианидов некоторых тяжелых металлов (никель, кобальт, цинк). Соответственно выделяют методы меркуриметрии, фторидометрии, цианидометрии. Кроме того, выделяют комплексонометрию, или комплексонометрическое титрование – метод, основанный на использовании реакций образования комплексонатов – комплексных соединений катионов металлов с комплексонами.
Неорганические однодентатные лиганды (ОН-, F-, CN-, NH3 и др.) ограниченно применяются в комплексометрии. Это связано с тем, что однодентатные лиганды реагируют с ионами металла с координационными числами больше единицы ступенчато, с образованием спектра

760
промежуточных соединений. Ступенчатые константы устойчивости промежуточных соединений близки друг к другу, вследствие чего скачки титрования на ТКТ, отвечающие отдельным ступеням реакции титрования, перекрывают друг друга. В результате получается ТКТ без скачков титрования, по которой невозможно подобрать индикатор, позволяющий регистрировать момент окончания образования какого-то конкретного комплексоната из спектра образующихся.
Этого недостатка лишены полидентатные лиганды с дентатностью больше пяти. Они с ионами металла реагируют в отношении 1: 1, вследствие этого соответствующая ТКТ имеет скачок титрования и по ней можно подобрать индикатор для регистрации ТЭ в реальном титровании.
Основным условием комплексометрического титрования является требование, предъявляемое к реакции, которая должна протекать таким образом, чтобы в точке эквивалентности определяемые катионы были практически полностью связаны в комплекс. Константа нестойкости таких комплексов должна быть очень малой величиной.
Кривые комплексометрического титрования (рисунок 4.6.5) обычно представляют собой зависимость рY = -lg[M] от объема титранта. До точки эквивалентности рассчитывается концентрация неоттитрованных ионов металла. В точке эквивалентности, которая соответствует скачку потенциала, фактически все ионы металла вошли в комплекс. После точки эквивалентности равновесную концентрацию ионов металла рассчитывают исходя из выражения для условной константы устойчивости.
Рисунок 4.6.5 – Кривые комплексометрического титрования
Скачок на кривой титрования зависит от:
-константы нестойкости комплекса (чем более устойчив комплекс, тем больше скачок титрования),
-концентрации реагентов (чем больше концентрация, тем больше
скачок),
761
- рН титруемого раствора.
Комплексометрическое титрование (комплексометрия или комплексонометрия) основанно на применении реакций образования прочных комплексных соединений катионов с органическими реактивами, называемыми комплексонами.
Комплексоны являются производными аминополикарбоновых кислот. Простейшим комплексоном, известным под названием комплексон I, служит трехосновная нитрилотриуксусная кислота (сокращенно H3Y). Наибольшее значение приобрела этилендиаминтетрауксусная кислота (ЭДТУ), комплексон II, четырехосновная кислота (сокращенно H4Y).
На практике обычно применяют динатриевую соль этилендиаминтетрауксусной кислоты, которую называют комплексоном III, ЭДТА, или трилон Б (сокращенно Na2H2Y). ЭДТА образует со многими катионами металлов устойчивые малодиссоциированные растворимые в воде внутрикомплексные соли. В комплексах часть связей носит ионный характер, часть – донорно-акцепторный. Трилон Б с ионами металлов любого заряда образует четырехпятиили шестикоординационный комплекс с пятичленными циклами. Атом металла находится в окружении атомов кислорода и атомов азота, находящихся в цис-положении.
Метод, в котором используют трилон Б, называют трилонометрией. Трилонометрический метод анализа основан на мгновенном образовании малодиссоциированных комплексных соединений различных катионов с трилоном Б. В водном растворе трилон Б диссоциирует и имеет кислую реакцию.
Nа2Н2Y = 2Nа+ + Н2Y2-
В реакциях комплексообразования реакции между трилоном Б и ионами металлов протекают стехиометрически в соотношении 1:1. Следовательно, молярная масса эквивалента ЭДТА и определяемого иона металла равны их молярным массам. При титровании ЭДТА солей металловкомплексообразователей протекают следующие реакции:
М2+ + Н2Y 2- → МY2- + 2 Н+ М3+ + Н2Y 2- → МY- + 2 Н+ М4+ + Н2Y 2- → МY + 2 Н+
Образующиеся комплексные соединения различаются лишь по заряду. Ионы водорода понижают рН раствора, в результате повышения кислотности среды требуемого комплексного соединения может не получиться. Поэтому титрование проводят в буферном растворе, поддерживающем определенное значение рН.
Для титриметрического определения необходимо знать точную концентрацию вещества. Стандартным называется раствор, для которого известна точная концентрация растворенного вещества.

762
Кривые комплексонометрического титрования имеют следующий вид (рисунок 4.6.6), содержат скачок титрования, по которому определяется точка эквивалентности. Принцип построения кривых комплексонометрического титрования такой же, как для комплексометрического титрования. До точки эквивалентности рассчитывается концентрация неоттитрованных ионов металла. В точке эквивалентности, которая соответствует скачку потенциала, фактически все ионы металла вошли в комплекс. После точки эквивалентности равновесную концентрацию ионов металла рассчитывают исходя из выражения для условной константы устойчивости.
Рисунок 4.6.6 – Кривые комплексонометрического титрования
Скачок на кривой титрования зависит от:
-константы нестойкости комплекса (чем более устойчив комплекс, тем больше скачок титрования),
-концентрации реагентов (чем больше концентрация, тем больше
скачок),
-рН титруемого раствора.
Комплексонометрическое титрование применяют для определения препаратов кальция, цинка, магния. Широко используют комплексонометрию при анализе воды для определения ее жесткости, которая вызвана присутствием солей кальция и магния. При этом можно определить как общую жесткость, используя индикатор эриохром черный Т или кислотный хромовый темно-синий, так и жесткость, обусловленную присутствием кальция, применив мурексид в качестве индикатора.
Комплексонометрическое титрование позволяет с высокой точностью проводить анализ различных сплавов и минералов. При этом возможно определение многих элементов при совместном присутствии, если использовать пригодные для этой цели металлохромные индикаторы и регулировать рН среды.
Метод комплексонометрического титрования точен, выполняется
763
быстро и просто и имеет высокую избирательность, что обеспечило широкое применение метода в практике химического анализа.
Конечную точку титрования устанавливают визуально по изменению окраски комплексонометрических индикаторов (металлоиндикаторов), а также потенциометрически, фотометрически, амперометрически или другими методами.
Металлоиндикаторы – это органические реагенты, обратимо изменяющие окраску под влиянием Меn+-ионов. Индикаторы комплексонометрии также образуют с ионами внутрикомплексные соли, которые по условиям титрования должны быть менее устойчивы по сравнению с комплексонатами ионов данного металла; причём цвет комплексонов должен отличаться от цвета свободного индикатора.
К металлоиндикаторам предъявляется ряд требований:
-металлоиндикаторы должны в выбранной области рН образовывать с ионами металлов устойчивые комплексы MInd;
-комплекс иона металла с индикатором должен быть кинетически лабильным и быстро разрушаться под действием титранта;
-изменение окраски в конечной точке титрования должно быть контрасным.
В качестве индикаторов в комплексонометрии применяют красители: мурексид, кислотный хром тёмно-синий, кислотный хромоген чёрный специальный (эрихром чёрный Т) и др. Последние два в щелочной среде имеют синюю окраску. Ионы кальция, магния и ряда других металлов образуют с индикаторами внутрикомплексные соединения (MeInd), окрашенные в вишнёво-красный цвет.
764
4.7 ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
ПЛАН
4.7.1 Электрохимические методы анализа.
4.7.1.1Потенциометрический метод анализа. 4.7.1.1.1 Общая характеристика метода.
4.7.1.1.2 Понятия и термины, используемые в потенциометрии. 4.7.1.1.3 Электроды в потенциометрии.
4.7.1.1.4 Прямая потенциометрия.
4.7.1.1.5 Потенциометрическое титрование.
4.7.1.2Вольтамперометрия.
4.7.1.2.1Полярография – основы метода.
4.7.1.2.2Качественный анализ.
4.7.1.2.3Количественный анализ.
4.7.1.2.4Амперометрическое титрование.
4.7.1.3Электрогравиметрический метод анализа.
4.7.1.4Кулонометрия.
4.7.1.4.1 Прямая кулонометрия (кулонометрия при постоянном контролируемом потенциале).
4.7.1.4.2 Кулонометрическое титрование (кулонометрия при контролируемой силе тока).
4.7.2 Оптические методы анализа.
4.7.2.1 Общая характеристика оптических методов анализа. 4.7.2.1.1 Электромагнитное излучение и его свойства. 4.7.2.1.2 Электромагнитный спектр.
4.7.2.1.3 Спектры атомов и их характеристики. 4.7.2.1.4 Оптические методы анализа. Классификация. 4.7.2.2 Абсорбционная спектроскопия.
4.7.2.2.1 Основной закон светопоглощения (закон Бугера – Ламберта –
Бера).
4.7.2.2.2Аппаратурное оформление метода.
4.7.2.2.3Фотометрический метод анализа.
4.7.2.2.4Фотометрическое титрование
4.7.2.3Использование спектров атомов в аналитических целях.
4.7.2.4Атомно-абсорбционный метод анализа.
4.7.2.5Эмиссионная фотометрия пламени.
4.7.2.6Люминесцентный метод анализа.
4.7.2.7Рефрактометрический метод анализа.
4.7.3 Хроматография.
4.7.3.1 Общие положения и понятия хроматографии.
4.7.3.2 Хроматограммы и основные принципы хроматографического разделения.
4.7.3.3 Хроматографический метод анализа.
765
Все методы количественного анализа основаны на изучении свойств вещества, связанных с концентрацией определенной зависимостью. В так называемых классических методах аналитической химии (гравиметрическом
ититриметрическом) в качестве таких свойств используются масса вещества
иобъем раствора. Однако вещество обладает совокупностью многих свойств, оно может поглощать и излучать свет, подвергаться радиоактивному распаду, прово-дить электрический ток и т.п. Поэтому классические методы постепенно уступают место физико-химическим методам (инструментальным) анализа.
Использование различных физических и физико-химических свойств вещества в аналитических целях лежит в основе физикохимических методов анализа.
Эти методы обладают многими существенными достоинствами:
1.Высокая чувствительность, которая позволяет легко проводить определения при очень малом содержании компонента (10-5 % и меньше). Некоторые методы настолько чувствительны, что позволяют считать чуть ли не отдельные атомы (измерения радиоактивности). В области малых концентраций классические методы вообще неприменимы, и анализ может быть выполнен только физико-химическими методами.
2.Экспрессность, т.е. быстрота получения результатов. Физикохимические методы успешно конкурируют с классическими и в области средних концентраций, так как даже приближенный результат анализа, полученный в течение нескольких минут, нередко является более ценным, чем самые точные данные, полученные через несколько часов или дней. Например, своевременная информация о составе сырья, о степени протекания процесса дает возможность технологу активно вмешиваться в ход технологического процесса и вводить необходимые коррективы.
3.Универсальность. В настоящее время можно провести анализ любого объекта, используя тот или иной метод анализа.
Физико-химические методы анализа позволяют проводить анализ на расстоянии (анализ лунного грунта, анализ морских вод на больших глубинах и т.д.).
Анализ с помощью некоторых физико-химических методов может быть выполнен без разрушения образца (недеструктивный анализ), что имеет большое значение для медицины, криминалистики, некоторых отраслей промышленности и т.д.
Часто практический интерес представляет так называемый локальный анализ, который позволяет провести определение компонента в данной «точке» образца, распределение элемента по поверхности анализируемого объекта. Этот анализ имеет особенно большое значение в минералогии, криминалистике, археологии, металловедении, где состав отдельных включений определяет качество материала.
4.Экономичность. Несмотря на некоторую дороговизну приборов, физико-химические методы анализа достаточно быстро окупают затраты, так
766
как сокращается время анализа, увеличивается производительность, сокращаются затраты на обслуживающий персонал и реактивы.
5. Возможность автоматизации. Использование электронных средств в аналитической химии является перспективным не только для расчета результатов анализа и статистической обработки, но и для решения других аналитических задач. С их помощью можно более надежно выделять аналитический сигнал, проводить более четкое разрешение перекрывающихся сигналов. Компьютеры, встроенные в аналитические приборы, значительно расширяют возможности этих приборов.
Все эти достоинства открывают перед физико-химическими методами самые широкие перспективы применения их в производстве, медицине, технике и науке.
Исключительное значение эти методы имеют для решения таких важных задач, как улучшение качества продуктов питания, лекарств, повышение эффективности производства, охрана окружающей среды, очистка питьевых и сточных вод, препаратов, получение продуктов высокой степени чистоты.
Погрешности анализа физико-химическими методами составляют в среднем 2–5 %, что несколько превышает погрешности классических методов анализа. Однако такое сравнение относится к разным концентрационным областям. При небольшом содержании определяемого компонента (10-3 % и менее) классические методы вообще непригодны, а при больших концентрациях физико-химические методы успешно конкурируют с ними, а некоторые даже превосходят классические методы по точности (например, кулонометрия). В настоящее время погрешность определений физико-химическими методами снижается за счет конструирования более точных приборов и совершенствования методик анализа.
Однако классические химические методы своего значения не потеряли. Они используются там, где при высоком содержании определяемых компонентов не требуются ограничения по времени проведения анализа.
Недостатком большинства физико-химических методов является то, что для их применения используются стандартные растворы, эталоны, а также часто градуировочные графики.
В группе физико-химических методов анализа иногда выделяют физические методы. Четкого и однозначного критерия для такого выделения нет, поэтому выделение физических методов в отдельную группу принципиального значения не имеет.
Общее число физико-химических методов анализа велико – оно составляет несколько десятков, но наибольшее практическое применение находят три группы.
Самые распространенные методы анализа: 1) оптические; 2)
электрохимические; 3) хроматографические.
Среди указанных трех групп самой обширной по числу методов и важной по практическому значению является группа оптических методов
767
анализа. Перечень групп является далеко неполным. Сюда не вошли многие методы (кинетические, радиометрические, масс-спектральные и др.), что, конечно, нельзя считать признаком их второстепенности.
Физико-химические методы анализа основаны на использовании физико-химического свойства вещества (аналитического сигнала) и нахождении его зависимости от природы вещества и содержания его в анализируемой пробе.
Почти во всех физико-химических методах анализа применяют два методических приема: метод прямых измерений и метод титрования (косвенных измерений).
Впрямых методах используется непосредственно зависимость аналитического сигнала от природы анализируемого вещества и его концентрации. Качественной характеристикой являются свойства, зависящие от природы вещества (длина волны в спектроскопии, потенциал полуволны в полярографии и др.), количественной характеристикой служит интенсивность сигнала (интенсивность спектральной линии в первом случае
исила диффузионного тока – во втором).
Вкосвенных методах (методах титрования) в ходе титрования измеряется интенсивность аналитического сигнала и строится кривая титрования в координатах: интенсивность сигнала – объем добавленного титранта (мл). Кривые титрования могут иметь различный вид, так как интенсивность аналитического сигнала может быть связана с активностью определяемого вещества, титранта или продукта реакции. По кривой титрования находится точка эквивалентности. Расчеты проводятся как при обычном титровании, используя закон эквивалентов.
4.7.1 Электрохимические методы анализа
К электрохимическим методам анализа относятся методы, основанные на измерении электрических параметров анализируемых систем (количества электричества, прошедшего через раствор; силы предельного диффузионного тока; электропроводности электролита; потенциала электрода, погруженного в исследуемый раствор, и др.), изменяющихся в результате определенных реакций.
Классификация электрохимических методов анализа: 1. По способу выполнения:
Прямые (ионометрия, кулонометрия, потенциометрия, полярография и
др.).
Косвенные (титриметрия с электрохимическими методами индикации). Инверсионные (инверсионная вольтамперометрия и др.).
2. По количеству вещества, участвующему в электродном процессе:
Все вещество участвует в электродном процессе (электрогравиметрия, прямая кулонометрия и др.).
Лишь незначительная доля вещества подвергается электропре-

768
вращению (полярография, вольтамперометрия, прямая потенциометрия и др.).
3. По измеряемому электрохимическому параметру (используются чаще всего, так как измеряемый электрохимический параметр является наиболее важным классификационным признаком) (таблица 4.7.1).
Таблица 4.7.1 – Классификация электрохимических методов анализа по измеряемому электрохимическому параметру
Наименование |
Функциональная |
Измеряемый параметр |
Разновидность |
|
метода |
зависимость |
|||
|
|
|||
|
|
|
|
|
|
|
ЭДС гальванического |
Прямая потенциометрия. |
|
Потенцио- |
|
элемента, состоящего из |
||
E=f(a) |
Потенциометрическое |
|||
метрия |
индикаторного электродаи |
|||
|
титрование |
|||
|
|
электрода сравнения |
||
|
|
|
||
|
|
|
|
|
Электро- |
|
Масса вещества, |
Классический |
|
Q=f(m) |
выделившегося на |
электроанализ. Внутренний |
||
гравиметрия |
||||
|
электроде |
электролиз |
||
|
|
|||
|
|
|
Прямая кулонометрия. |
|
Кулонометрия |
Q=f(с) |
Количество электричества |
Кулонометрическое |
|
|
|
|
титрование |
|
Кондукто- |
|
|
Прямая кондуктометрия. |
|
λ=f(m) |
Электропроводность |
Кондуктометрическое |
||
метрия |
||||
|
|
титрование |
||
|
|
|
||
|
|
|
Полярография (Hg |
|
Вольтампе- |
I=f(E) |
Предельный ток. |
капающий электрод). |
|
I=f(С) при |
Амперометрия. |
|||
рометрия |
Предельный ток |
|||
Е=const |
Амперометрическое |
|||
|
|
|||
|
|
|
титрование |
4.7.1.1 Потенциометрический метод анализа 4.7.1.1.1 Общая характеристика метода
Потенциометрия основана на измерении потенциала, возникающего между электродом и раствором (или, точнее, ЭДС гальванического элемента), величина которого зависит от количества (концен-трации) или качества (природы) определяемого компонента. Такой электрод называют индикаторным. Таким образом, потенциал электрода в потенциометрии – аналитический сигнал.
Достоинства метода:
-Простота аппаратурного оформления.
-Быстрота проведения измерений.
-Высокая чувствительность.
-Возможность работы с малыми объемами.
-Большой диапазон концентраций.
-Универсальность. Метод позволяет использовать реакции нейтрализации, осаждения, комплексообразования, окислениявосстановления.

769
- Для анализа можно использовать мутные и окрашенные растворы.
По назначению потенциометрический метод может быть классифицирован как прямая потенциометрия и потенциометрическое титрование.
В потенциометрии выделяют две группы методов, различающиеся природой используемых индикаторных электродов и электрохимической реакцией на них:
1. Редоксметрия (классическая потенциометрия) с использованием электродов с электронным типом проводимости.
Возникающий при этом потенциал подчиняется уравнению Нернста:
(4.7.1)
где R – универсальная газовая постоянная, 8,31 Дж/моль; Т – температура, К;
F – число Фарадея, 96500 Кл/моль;
n – число электронов, участвующих в редокс-процессе; аMen+ – активность потенциалопределяющих ионов. Электрохимическая реакция, протекающая на электроде:
Меn+ + nе → Me0.
2. Ионометрия – разновидность потенциометрии, где в качестве индикаторных электродов используются электроды с ионным типом проводимости – мембранные ионселективные электроды (ИСЭ), потенциал которых возникает вследствие обмена ионами мембраны с раствором и подчиняется уравнению Никольского:
(4.7.2)
где Е0 – условный стандартный потенциал; аi – активность определяемых ионов i; аj – активность мешающих j-ионов; Kij – коэффициент селективности.
Знак «+» ставится при расчетах потенциалов катионселективных электродов, знак «-» – анионселективных.
Частный случай – рН-метрия.
Метод характеризуется сравнительно невысокой точностью определения. Потенциал измеряется на приборах с точностью ±1 мВ, что соответствует погрешности определения концентрации однозарядных ионов ±4%. Концентрации многозарядных ионов определяются с еще большими погрешностями.
Другим ограничением является пропорциональность величины
770
потенциала логарифму активности, а не концентрации, переход к концентрации затруднителен.
Существующие приемы ионометрических измерений направлены на преодоление указанных ограничений.
4.7.1.1.2 Понятия и термины, используемые в потенциометрии
1.Электроды – совокупность 2, 3 или 4-х контактирующих фаз, на границах которых возникают скачки электрических потенциалов.
Контактирующими фазами являются электролиты, металлы или полупроводники.
Если в контакте находятся расплавы или растворы электролитов, то такую систему электродом не называют.
2.Электрохимическая ячейка. Состоит из пары электродов,
погруженных в один общий или два разных электролита, находящихся в непосредственном контакте
М1|М1Х|М2Х|М2|М1
или соединенных солевым мостиком
М1|М1Х||М2Х|М2|М1.
Вертикальная черта показывает границу раздела, на которой может возникнуть скачок потенциала. Солевой мостик обозначают ||.
Электрохимические ячейки, в которых электрическая энергия образуется за счет химической реакции, называют гальваническими элементами.
3. Мембранная ячейка – ячейка, состоящая из мембранного электрода и двух электродов с металлической основой (электроды сравнения)
М1|М1Х(m1) | мембрана | М2Х(m2)|М1,
где m1 и m2 – моляльные концентрации соли в растворах.
4. Электродвижущая сила – разность электродных потенциалов правого е1 и левого е концов правильно разомкнутого гальванического элемента.
В потенциометрии используют только правильно разомкнутые гальванические элементы. Неправильно разомкнутый элемент обозначают
М1|М1Х||М2Х|М2.
Электродвижущая сила гальванического элемента равна разности потенциалов электродов исследуемой цепи.
Измерив величину ЭДС и зная значение потенциала одного из
771
электродов относительно водородного (стандартного электрода), можно определить потенциал другого электрода.
Далее по уравнению Нернста можно определить концентрацию вещества в растворе.
5.Электродная реакция (полуреакция) – реакция, протекающая на границе раздела фаз, составляющих электрод.
6.Солевой мостик (электролитический ключ) – трубка, наполненная концентрированным раствором электролита и погруженного своими концами
вдва других электролита, являющихся основными частями электрохимической ячейки.
7.Электродный потенциал (обычно условный потенциал) – ЭДС гальванического элемента, состоящего из стандартного водородного и исследуемого электродов.
Стандартный электродный потенциал – когда активность каждого компонента равна 1 моль/л.
Формальный электродный потенциал – когда концентрация каждого компонента равна 1 моль/л.
Мембранный потенциал – разность скачков электродных потенциалов на двух границах мембраны с двумя растворами – исследумым и внутренним.
4.7.1.1.3 Электроды в потенциометрии
В потенциометрии используют электроды двух типов:
1.Электроды сравнения – полуэлементы, потенциалы которых известны, постоянны и не зависят от состава изучаемого раствора.
2.Индикаторные электроды – полуэлементы, потенциалы которых зависят от активности (концентрации) определяемого компонента. Используются в сочетании с электродами сравнения. Применяются для измерения обратимых потенциалов исследуемых электродных реакций.
Электроды сравнения. Водородный электрод (с.в.э.) –
универсальный электрод сравнения. Представляет собой равновесную систему, состоящую из платиновой пластинки, покрытой платиновой чернью, погруженной в раствор, насыщенный водородом и содержащий ионы водорода.
Электрохимическую реакцию в данном случае можно представить, как:
2Н+ + 2е → Н2↑.
По международному соглашению принято считать потенциал с.в.э. при любой t0 равным 0 и относительно него выражать стандартные потенциалы других систем.
С.в.э. может использоваться и как индикаторный электрод для определения рН.
Однако возможности его применения ограничены, поэтому в качестве электродов сравнения могут выступать хлоридсеребряный (рассмотрен

772
ниже), каломельный и другие.
Индикаторные электроды. Согласно электрохимическим реакциям электроды, используемые в потенциометрии, различают по роду.
1. Электроды первого рода (металлические) – используют при измерении потенциала в качестве индикаторных.
Активные электроды 1-го рода – это металлическая пластинка или проволока, погруженная в раствор хорошо растворимой соли данного металла. Электроды 1-го рода обратимы относительно собственных ионов, а их потенциал – функция активности этих ионов в растворе. Например, серебряный электрод (Ag|Ag+): электрохимическая реакция:
Ag+ + е → Ag°.
Электрод активен и сам принимает участие в обмене электрона. Потенциал такого электрода подчиняется уравнению Нернста:
К этой группе электродов относят и индифферентные электроды, которые не участвуют в электрохимической реакции, а только обеспечивают перенос электронов для окислительно-восстановительной реакции, протекающей в растворе.
Эти электроды представляют собой пластинку или проволоку, изготовленную из инертных металлов (Pt, Au и др.), погруженную в раствор, содержащий сопряженную редокс-пару, например, платиновый электрод. Платина в раствор ионов не поставляет, а выступает лишь переносчиком электронов от окислителя к восстановителю в растворе.
Потенциал электрода зависит от активности окисленной и восстановленной форм. Например, потенциал платинового электрода, погруженного в раствор, содержащий Fe3+ и Fe2+:
2. Электроды второго рода – это системы, в которых металл электрода покрыт труднорастворимой солью этого металла и находится в растворе, содержащим хорошо растворимую соль с теми же анионами, т.е. это металл, находящийся в равновесии с его малорастворимой солью. Например, хлоридсеребряный электрод (х.с.э.) Ag|AgCl|Cl‾ (рисунок 4.7.1).

773
Рисунок 4.7.1 – Хлоридсеребряный электрод сравнения: 1 – асбестовое волокно, обеспечивающее контакт с анализируемым раствором, 2 – внешний раствор KCl (насыщенный); 3 – отверстие для контакта; 4 – внутренний раствор KCl (насыщенный); 5 – отверстие для ввода раствора KCl
Электрохимическая реакция: AgCl + e → Ag0 + Сl‾. Потенциал такого электрода зависит от активности аниона соли, хотя сами анионы при этом не окисляются и не восстанавливаются:
Потенциалы электродов 2-го рода хорошо воспроизводимы и устойчивы. Такие электроды чаще используют в качестве электродов сравнения.
Электроды 2-го рода используют и как индикаторные для определения концентрации собственных ионов. При 25 оС потенциал х.с.э. равен 0,222 В по отношению к водородному электроду:
3. Ионселективные (мембранные) – сенсоры (чувствительные элементы), потенциал которых линейно зависит от lg a определяемого иона. Среди таких электродов наиболее часто используется стеклянный электрод (рисунок 4.7.2).
Высокое содержание Na+ в мембране способствует обмену между ионами Na+ из мембраны и H+ из раствора.
Потенциал стеклянного электрода зависит от активности ионов H+ в растворе и определяется:
(4.7.3)
где К – константа, зависящая от сорта стекла и устройства электрода.

774
Рисунок 4.7.2 – Устройство стеклянного электрода: 1– тонкая рH-чувствительная стеклянная мембрана, заполненная 0,1М раствором HCl (2); 3 – внутренний электрод: серебряная проволока, покрытая труднорастворимой солью AgCl; 4 – стеклянная трубка; 5 – изоляция; 6 – токоотвод
Преимущества стеклянного электрода:
-Электрохимическое равновесие устанавливается мгновенно.
-Электрод не адсорбирует ПАВ.
-Отсутствуют влияния окислителей и восстановителей на работу электрода.
-Электрод применим в широком диапазоне рН. Не рекомендуется использовать электрод при рН больше 10, так как нарушаются его функции.
С другим составом стекла электроды селективны на другие ионы, например, существуют pNa-, pK-, pCa-, pMg-, pNO3-, pCl- и другие стеклянные электроды.
Существуют жидкие мембранные электроды, которые представ-ляют собой две несмешивающиеся с водой жидкие фазы.
Измерительное устройство, применяемое для измерения потен-циала – потенциометр. Такие приборы заводского типа называют рН-метрами (иономерами), поскольку они предназначены для измерения потенциалов ячеек, содержащих рН-чувствительный стеклянный электрод с высоким сопротивлением. Шкала этих приборов калибру-ется как в милливольтах, так
ив единицах рН.
4.7.1.1.4 Прямая потенциометрия
Прямая потенциометрия основана на применении уравнения Нернста для определения концентрации (активности) по экспериментально установленному потенциалу электрода. Метод основан на точном измерении электродного потенциала Еравн и нахождении по уравнению Нернста концентрации (активности) потенциалопреде-ляющего иона в растворе:

775
(4.7.4)
где E0 – стандартный (нормальный ) потенциал электрода при концентрации определенных ионов 1моль экв/л (справочная величина);
R – универсальная газовая постоянная, равная 8,314 Дж/(град.моль); Т– абсолютная температура, °К;
n – заряд иона;
F – постоянная Фарадея 96500 Кл/моль;
аOx и аRed – активности окисленной и восстановленной формы вещества в растворе.
Перейдя от натурального логарифма к десятичному и подставив численные значения констант, получим
(4.7.5)
Этот метод является, пожалуй, единственным, позволяющим непосредственно в растворе определять активности ионов.
В ионометрии в качестве индикаторных применяют обычно мембранные ионселективные электроды, потенциал которых зависит от концентрации определяемых ионов. Электродами сравнения служат электроды 2-го рода (например, хлоридсеребряный).
Концентрацию определяемых ионов находят одним из методов количественного анализа:
1.Метод градуировочного графика E = f(C).
2.Метод добавок (анализ сложных по составу и сильно разбавленных растворов).
3.Метод градуировки электрода по растворам с точно известной концентрацией определяемых ионов (буферные растворы) – ионометрия.
Метод градуировочного графика:
1.Для построения градуировочного графика в координатах E – lgCM (E
–pCM) измеряют ЭДС элемента при нескольких концен-трациях определяемого иона и постоянной ионной силе.
2.По величине ЭДС и градуировочному графику находят концентрацию определяемого компонента в растворе (рисунок 4.7.3).

776
Рисунок 4.7.3 – Градуировочный график для определения концентрации методом прямой потенциометрии
Поскольку градуировочный график представляет собой прямую, то произвести расчеты по найденному потенциалу несложно.
Важной особенностью метода градуировочного графика является необходимость постоянства условий проведения измерений. При проведении измерений следует прежде всего уделять внимание уравниванию температуры и ионной силы, как в стандартных растворах, так и в анализируемых пробах. Несоблюдение этого условия ведет к увеличению погрешности измерений.
4.7.1.1.5 Потенциометрическое титрование
Потенциометрическое титрование относится к косвенным методам анализа. Метод основан на установлении точки эквивалентности по резкому изменению потенциала индикаторного электрода при титровании.
В основе метода лежит вариант классического титрования, но не индикаторной, а с потенциометрической индикацией точки эквивалентности с помощью индикаторного и вспомогательного электродов.
Титрант добавляют точно известными порциями и записывают показания прибора Е. По результатам титрования строят интегральную (Е – V, см3), или дифференциальную (∆Е/∆V – V, см3) кривые титрования (рисунок 4.7.4).
В простом и удобном методе Грана точка эквивалентности определяется по графику в координатах ∆Е/∆V – V (рисунок 4.7.4, г). Перед точкой эквивалентности и после нее кривая линейна, а сама точка эквивалентности находится как точка пересечения этих прямых.


778
сравнения, может быть представлен схемой:
Потенциометрическая ячейка для измерения потенциала с ионселективным электродом в качестве индикаторного представлена на рисунке
4.7.5.
Рисунок 4.7.5 – Схема установки для измерения потенциала с ионселективным электродом: 1 – вспомогательный электрод ЭВЛ-1МЗ; 2 – наконечник
ионселективного электрода; 3 –жидкая мембрана; 4 – магнитная мешалка; 5 – магнит
Методика потенциометрического титрования. В стакан для титрования емкостью 50–100 мл переносят пипеткой аликвотную часть испытуемого раствора. Прибавляют необходимые реагенты и погружают в раствор электроды. Если уровень раствора мал и электроды недостаточно погружены в раствор, добавляют нужное количество воды (или другого растворителя), включают мешалку и титруют, записывая показания прибора. Целесообразно первое титрование выполнить ориентировочно, прибавляя титрант пор-цими по 1–2 мл.
Результаты измерений помещают в таблицу, где наряду с исходными данными вносят и данные для расчетов по дифференциальным кривым (таблица 4.7.3).
Для построения интегральной кривой используются данные столбцов 1 и 2, для построения дифференциальной кривой – столбцов 1 и 5, для построения кривой второй производной – столбцов 1 и 7.

779
Таблица 4.7.3 – Результаты потенциометрического титрования
Построив график зависимости Е от объема титранта, находят положение скачка титрования и при повторном титровании вначале добавляют титрант относительно большими порциями (2–5 мл), а вблизи скачка титрант добавляют по 0,1–0,05 мл или даже по одной капле. Точке эквивалентности соответствует область максимального скачка потенциала, максимум первой производной от потенциала по объему титранта или момент резкого падения второй производной. Графическое нахождение точки эквивалентности показано на рисунке.
Расчет результатов потенциометрического титрования проводят так же, как в классической титриметрии.
Практическое применение метода:
-В анализе объектов окружающей среды и технологических растворов (определение рН и концентрации ионов).
-В медицине и других биологических исследованиях (например, Саэлектрод позволяет определять жесткость воды, содержание Са в биологических объектах и др.).
-Для изучения равновесий в растворах: определения констант равновесия, констант диссоциации, констант устойчивости комплексных соединений, ПР.
4.7.1.2 Вольтамперометрия
Вольтамперометрия основана на изучении поляризационных или
вольтамперных кривых (кривых зависимости силы тока от напряжения), которые получаются, если при электролизе раствора ана-лизируемого вещества постепенно повышать напряжение и фиксиро-вать при этом силу тока.
Основатель метода – чешский ученый Я. Гейровский (1922 г.), за что он в 1959 г. получил Нобелевскую премию.
Гейровский проводил электролиз на Hg-капающем электроде. Вольтамперометрию, связанную с использованием ртутного капающего электрода, стали называть полярографией. Таким образом, в полярографии поляризуемый электрод (индикаторный) – Hg-капающий с постоянно обновляющейся поверхностью (катод), а электрод сравнения – слой ртути на дне ячейки (анод).
4.7.1.2.1 Полярография – основы метода
Рассмотрим электролиз в системе, где катодом служит Hg-капающий электрод, а анодом является практически неполяризуемый каломельный
780
электрод.
Если в растворе нет веществ, способных восстанавливаться под действием электрического тока, сила тока I будет пропорциональна приложенному напряжению E (закон Ома):
I = E/R.
(4.7.10)
В присутствии веществ, способных восстанавливаться, вид кривой изменится.
1.По достижении потенциала восстановления ионы начнут разряжаться на Hg-электроде.
2.Концентрация ионов у поверхности ртутной капли начнет уменьшаться, а сила тока возрастать.
3.Однако за счет диффузии к поверхности капли доставляются новые порции ионов. Сила тока в цепи будет зависеть от скорости диффузии,
которая пропорциональна разности концентраций в массе раствора СМ и в приэлектродном слое CМ0.
I = k(СМ - CМ0). |
(4.7.11) |
При некотором потенциале концентрация ионов у поверхности ртутной капли сильно уменьшится и скорость разряда ионов станет равной скорости диффузии.
Концентрация ионов в массе раствора будет постоянна, а концентрация у поверхности электрода близка к 0, поэтому их разность будет постоянной. Наступившее состояние равновесия будет харак-теризоваться постоянной силой тока, не изменяющейся при дальней-шем увеличении напряжения.
Этот постоянный ток, контролируемый диффузией, называют
диффузионным током (Id).
Связь диффузионного тока Id с концентрацией иона СМ передается уравнением Ильковича:
Id = 605 z D1/2m2/3t1/6CM,
(4.7.12)
где
z – заряд иона;
D – коэффициент диффузии, см2/с;
m – масса ртути, вытекающей из капилляра мг/с;
t – время образования ртутной капли (период капания), с; C – концентрация деполяризатора в растворе, ммоль/л.
781
При постоянных условиях полярографирования уравнение приобретает
вид
Id = kCM,
(4.7.13)
где k – константа, зависящая от природы определяемого иона, среды, температуры и характеристик электрода. Поэтому в работах по полярографии всегда указывается характеристика капилляра.
Линейная зависимость I от CM является основой количественного полярографического анализа.
Зависимость силы тока от приложенного напряжения при обратимом электродном процессе передает уравнение
(4.7.14)
где Е – потенциал в любой точке волны, I – сила тока в этой точке волны,
Е1/2 – потенциал полуволны;
R – универсальная газовая постоянная, равная 8,314Дж/(град.моль); T – абсолютная температура, °К; n – заряд иона;
F – постоянная Фарадея, 96500 Кл/моль.
Это уравнение называется уравнением полярографической волны, а величина E1/2 называется потенциалом полуволны.
Типичная полярографическая волна (полярограмма) приведена на рисунке 4.7.6.
Участок I от начала регистрации электрохимического сигнала до начала электрохимической реакции – это остаточный ток, его величина имеет порядок 10-7 А. Участок II – резкое увеличение I от Е за счет электрохимической реакции. Так как на катоде начинается раз-ряд ионов, сила тока резко возрастает, стремясь к предельной величине диффузионного тока Id. Когда I станет равной Id, уравнение полярографической волны приобретает вид Е = Е1/2. Участок III – диффузионный ток, достигнув предельного значения, остается постоянным, реакция завершена.

782
Рисунок 4.7.6 – Типичный вид полярографической волны
Таким образом, потенциал полуволны Е1/2 не зависит от силы тока и концентрации восстанавливающегося иона и является качественной характеристикой иона в растворе данного фонового электролита.
Фоновый электролит (обычно раствор соли щелочного или щелочноземельного металла) добавляют в раствор для обеспечения высокой электропроводности раствора и предотвращения недиффузи-онных процессов (например, миграции ионов).
Если в растворе присутствуют несколько веществ, потенциалы полуволны которых различаются на 100 мВ и больше, то на полярограмме будет не одна волна, а несколько (рисунке 4.7.7).
Рисунок 4.7.7 – Полярограмма раствора, содержащего цинк, никель и кадмий
Схема полярогафической установки представлена на рисунке 4.7.8.
Анализируемый раствор, находящийся в электролизере 1, на дне которого имеется слой ртути, является анодом. Часто в качестве анода


784
Для идентификации неизвестного вещества найденный потенциал полуволны сравнивают с табличными значениями.
4.7.1.2.3 Количественный анализ
Количественный анализ основан на пропорциональной зависимости между силой предельного диффузионного тока I (мка) и концентрацией деполяризатора С.
I = kC |
(4.7.15) |
На практике для количественных определений уравнение Ильковича, как правило, не используют, поскольку определение численных значений всех его параметров – слишком трудоемкая работа. Чаще всего для целей количественного анализа пользуются высотой полярографической волны h, выраженной в мм.
Для определения концентрации компонента используют методы: градуировочного графика; стандартных растворов; добавок (расчетный или графический вариант).
Метод градуировочного графика. Метод градуировочного графика,
построенного в координатах Id – С, используют наиболее часто. Он представляет собой прямую линию, проходящую через начало координат
(рисунок 4.7.10).
Рисунок 4.7.10 – Градуировочный график в полярографическом анализе
Метод обычно применяют при выполнении серийных анализов. При построении градуировочного графика часто вместо величины предельного диффузионного тока откладывают просто высоту полярографической волны h, измеренной на полярограмме в миллиметрах (рисунок 4.7.11).

785
Рисунок 4.7.11 – Типичный вид полярограмм определяемого элемента при концентрациях С1 ‹ С2 ‹ С3
Для построения градуировочного графика полярографируют несколько стандартных растворов. Измеряют для каждого раствора высоту полярографической волны h (рисунок). Строят график зависимости высоты полярографической волны, которая пропорциональна силе диффузионного тока, от концентрации анализируемого вещества. В соответствии с уравнением Ильковича график представляет собой прямую, проходящую через начало координат. Затем полярографируют анализируемый раствор и по графику находят концентрацию компонента.
Метод стандартных растворов. При выполнении небольших серий анализов находит применение метод сравнения со стандартными растворами. В этом методе в одних и тех же условиях снимают полярограммы стандартного и анализируемого растворов, измеряют высоты полярограмм и из пропорции, основанной на уравнении Ильковича, рассчитывают неизвестную концентрацию Сх по уравнению
(4.7.16)
где, Сст – концентрация стандартного раствора;
hx и hст – высота волны анализируемого и стандартного растворов.
Метод добавок
При анализе растворов, качественный состав которых точно неизвестен, обычно применяют метод добавок стандартного раствора. Его сущность состоит в том, что вначале снимают полярограмму анализируемого раствора и на ней определяют величину предельного диффузионного тока Id:
I = kC.
После этого к анализируемому раствору добавляют известное количество стандартного раствора, снимают полярограмму этого раствора и

786
на ней определяют величину предельного диффузионного тока Iх+ст, который равен:
Iх+ст = k(Cх + Сст).
(4.7.17)
Если разделить эти уравнения друг на друга, получим
(4.7.18)
Откуда
(4.7.19)
4.7.1.2.4 Амперометрическое титрование
Метод более универсален, чем прямая вольтамперометрия, так как определяемое вещество не обязательно должно быть электроактивным. Важным является необходимость окисления или восстановления на электроде хотя бы одного из двух участников реакции или продукта их взаимодействия. Электроактивным может быть титрант или продукт реакции.
Впроцессе титрования после прибавления отдельных порций реагента измеряют силу тока при напряжении, соответствующем величине предельного тока. Кривые амперометрического титрования строятся в координатах сила тока – объем титранта и графически находят точку эквивалентности. Кривая состоит из двух участков, пересечение которых соответствует точке эквивалентности (рисунок).
Вид кривой амперометрического титрования зависит от того, какой компонент реакции вступает в электродную реакцию. Поэтому кривые бывают разных видов (рисунок 4.7.12).
Втом случае, когда электрохимически активный компонент в реакции титрования отсутствует, для амперометрического титрования используют полярографические индикаторы. Полярографический (или амперометрический) индикатор представляет собой вещество, способное электрохимически окисляться или восстанавливаться, но оно взаимодействует с титрантом значительно слабее анализируемого вещества. Это приводит к тому, что концентрация полярографического индикатора, а следовательно и сила диффузионного тока начнут уменьшаться только после точки эквивалентности (рисунок е).

787
Рисунок 4.7.12 – Кривые амперометрического титрования: а – электроактивно определяемое вещество; б – электроактивен титрант; в – электроактивны определяемое вещество и титрант; г – на электроде одно вещество восстанавливается, a другое – окисляется; д – электроактивен продукт реакции; е – используется полярографический индикатор
Условия выполнения анализа:
1. Титрование проводят при постоянном потенциале, соответствующем предельному диффузионному току деполяризатора
(Е=const).
2.В раствор добавляют фоновый электролит, природу и концентрацию которого подбирают предварительно.
3.Для регистрации аналитического сигнала необходима система двух электродов – рабочего (индикаторного, поляризуемого) микроэлектрода и электрода сравнения (неполяризуемого).
4.Скорость перемешивания раствора должна быть постоянной.
Схема установки для амперометрического титрования представлена на рисунке 4.7.13.

788
Pисунке 4.7.13 – Cхема установки для амперометрического титрования с двумя индикаторными электродами: 1 – платиновые электроды; 2 – стакан для титрования;
3 – мешалка; 4 – бюретка; 5 – гальванометр
Ha индикаторный электрод подают такое напряжение, при котором через раствор исследуемого вещества, титранта и продукта их реакции течет предельный диффузионный ток. Tак как его величина пропорциональна концентрации электроактивного вещества, то по мере изменения концентрации в ходе титрования меняется и величина предельного диффузионного тока.
Строят график зависимости силы тока от объема, израсходованного титранта. Пересечение двух ветвей кривой титрования соответствует конечной точке (рисунок).
Амперометрическое титрование находит применение в различных отраслях, особенно в химической и фармацевтической промышленности. Амперометрическое титрование более экспрессно, селективно и чувствительно, чем потенциометрическое. Метод амперометрического титрования предпочтителен при определении низких концентраций (до 10-5 моль/дм3). Точность анализа его может составлять 0,1 %.
4.7.1.3 Электрогравиметрический метод анализа
В основе метода – процесс электролиза.
Электролизом называется химическое разложение вещества под действием электрического тока.
На катоде (- заряженном электроде) происходит восстановление. На аноде (+ заряженном электроде) происходит окисление.
Основные законы электролиза установлены Фарадеем:
1.Масса вещества, выделившегося при электролизе, пропорциональна количеству электричества, прошедшего через раствор.
2.При прохождении через раствор одного и того же количества электричества на электродах выделяется одно и то же количество ве-щества эквивалента.

789
Законы электролиза:
(4.7.20)
где m – масса вещества, выделившегося при электролизе, г; Q – количество электричества, Кл;
М – молярная масса эквивалента вещества, моль/л; I – сила тока, А;
t – время электролиза, с;
96500 – число Фарадея, равное количеству электричества для выделения 1 моля эквивалента вещества, Кл.
Сущность метода. Анализируемое вещество количественно выделяют из раствора электролизом и по массе его на электроде рассчитывают содержание определяемого элемента в пробе.
Схема установки для проведения электролиза показана на рисунке
4.7.14.
Рисунок 4.7.14 – Схема установки для электрогравиметрических измерений
Для получения постоянного тока обычно используют выпрямитель переменного тока или батарею аккумуляторов 1. Скользящий контакт 2 позволяет регулировать подаваемое напряжение, которое измеряют вольтметром. Сила тока контролируется амперметром. При выделении металлов катод 5 обычно изготовляют из платиновой сетки, анод 4 – из платиновой спирали или пластинки. При выделении оксидов знаки электродов меняются: платиновая сетка становится анодом, а спираль – катодом. Раствор перемешивается механической или магнитной мешалкой 3.
790
Важнейшими требованиями к форме осаждения являются ее малая растворимость и чистота, т.е. отсутствие загрязнений. Эти требования в электрогравиметрическом методе выполняются идеально, так как большинство металлов и указанные оксиды практически не растворимы в воде, а при электролитическом выделении металлов или оксидов соосаждение почти всегда можно предупредить путем подбора условий электролиза. Полученный осадок металла или оксида удобен для промывания и взвешивания.
Внутренний электролиз. В методе внутреннего электролиза внешнего источника тока не требуется. Здесь используется способность металлов с более положительным электродным потенциалом выделяться в свободном виде из растворов их солей под действием металлов с меньшим значением стандартного потенциала. Пластинка менее благородного металла, являющаяся анодом, соединяется платиновым катодом и, таким образом, выделение анализируемого благородного металла происходит на платине. При небольшом содержании определяемого элемента осаждение металла на платиновом катоде происходит без каких-либо осложнений, но при больших концентрациях наряду с осаждением на катоде может происходить некоторое выделение металла на аноде. Чтобы исключить этот процесс, анод покрывают тонкой коллоидной пленкой или катодное и анодное пространство разделяют пористой перегородкой.
Одним из важных достоинств метода внутреннего электролиза является возможность проведения тонких химических разделений, так как на платиновом катоде выделяются металлы только более благородные, чем металл анода.
4.7.1.4 Кулонометрия
Метод основан на определении количества электричества, которое расходуется в ходе электрохимической реакции.
Различают:
1.Прямую кулонометрию (все вещество подвергается электрохимическому превращению).
2.Кулонометрическое титрование (определяемое вещество реагирует
ститрантом, который генерируется в ячейке при электролизе специально подобранного раствора).
Однако какой бы вид кулонометрии не использовался, всегда должно выполняться условие: электрохимическому восстановлению или окислению должно подвергаться только анализируемое вещество со 100 % выходом по току.
4.7.1.4.1 Прямая кулонометрия (кулонометрия при постоянном контролируемом потенциале)
Метод прямой кулонометрии основан на непосредственном окислении или восстановлении анализируемого вещества на рабочем электроде,
791
исключающем прохождение побочных электрохимических реакций.
Прямая кулонометрия может быть выполнена в одном из следующих режимов:
1.При постоянном потенциале рабочего электрода в течение всего времени электролиза – такой режим называют потенциостатическим.
2.При постоянной силе тока в течение всего времени электролиза – режим называют амперостатическим.
В методе прямой кулонометрии в потенциостатическом режиме сила тока в течение всего времени электролиза непрерывно уменьшается, так как происходит уменьшение концентрации анализируемого вещества. Электролиз заканчивают при уменьшении силы тока практически до нуля.
Количество электричества, затраченное на анализ, измеряют либо с помощью кулонометров, либо графически, по построенной диаграмме в координатах: время электролиза – сила тока.
При выполнении кулонометрического анализа в амперометрическом режиме сила тока на протяжении всего времени электролиза поддерживается постоянной. Количество электричества в этом случае легко рассчитывается по уравнению
Q = I • t. (4.7.21)
Этот режим более экспрессный, чем потенциостатический, но его можно применять лишь в том случае, если есть возможность установить момент, когда электролиз анализируемого вещества полностью завершен.
Массу определяемого вещества рассчитывают по формуле
(4.7.22)
Методом прямой кулонометрии определяют ионы меди, свинца, висмута, мышьяка, урана и других металлов. Этот метод нашел применение также для анализа органических соединений, в том числе и лекарственных препаратов (аскорбиновой кислоты, новокаина, пикриновой кислоты, оксихинолина и др.).
Метод прямой кулонометрии очень чувствителен. Им можно определить до 10-9 г вещества в пробе. Ошибка определений не превышает
0,02 %.
4.7.1.4.2 Кулонометрическое титрование (кулонометрия при контролируемой силе тока)
Кулонометрическое титрование основано на электрохимическом получении титранта (электрогенерировании титранта) с последующей реакцией его с анализируемым веществом.
792
Если титрант электрогенерируется непосредственно в растворе анализируемого вещества, то такое титрование называется кулонометрическим титрованием с внутренней генерацией.
Если титрант получают электрогенерированием в отдельном сосуде, а затем подают его в анализируемый раствор, то такое титрование называют кулонометрическим титрованием с внешней генерацией. Этот вид титрования используется очень редко.
Кулонометрическое титрование всегда проводят в амперостати-ческом режиме и применяют большие токи для электролиза, что позволяет выполнять кулонометрическое титрование экспрессно. Поэтому используют установки с постоянной силой тока.
Так как титрант генерируется в количестве, точно эквивалентном содержанию анализируемого вещества, то по количеству электричества, израсходованного на генерацию титранта, можно рассчитать содержание определяемого вещества.
Достоинство кулонометрического титрования и преимущество перед обычным титрованием. Рабочий раствор не готовят и не стандартизируют. Титрант генерируется электрохимически непосредственно в присутствии анализируемого вещества и в количестве, необходимом только для данного титрования. Это позволяет использовать для титрования малоустойчивые и летучие вещества.
Кулонометрически может быть выполнен любой вид титрования: кислотно-основное, осадительное, комплексонометрическое, окислительновосстановительное.
Метод кулонометрического титрования по точности и чувствительности превосходит другие методы титрования. Он пригоден для титрования очень разбавленных растворов концентрацией до 10-6 моль/дм3, а погрешность определений не превышает 0,1–0,05 %.
4.7.2 Оптические методы анализа 4.7.2.1 Общая характеристика оптических методов анализа
Оптические методы анализа основаны на измерении эффектов взаимодействия вещества с электромагнитным излучением.
4.7.2.1.1 Электромагнитное излучение и его свойства
Электромагнитное излучение представляет собой вид энергии, которая распространяется с огромной скоростью.
Эта энергия существует в различных формах, из которых наиболее легко распознаются свет и тепло.
Распространение электромагнитного излучения удобнее представить в виде волнового процесса, характеризующегося такими параметрами, как скорость, частота, длина и амплитуда волны. В отличие от других волновых процессов, например, звука, для передачи электромагнитного излучения не нужна проводящая среда, оно легко распространяется в вакууме.
Но для объяснения явлений, связанных с поглощением или излучением

793
энергии, необходимо представлять излучение и в виде потока дискретных частиц, называемых фотонами.
Электромагнитное излучение удобно представить в виде электрического силового поля, колеблющегося перпендикулярно направлению распространения волны.
Волновые свойства электромагнитного излучения:
1. λ – длина волны – расстояние между двумя последующими максимумами или минимумами волны. Измеряется в нанометрах (нм), ангстремах (Å) или микрометрах (мкм).
1нм=10-9м
1Å=10-10м
1мкм=10-6м
2. Р – период излучения – время, необходимое для прохождения каждого последующего максимума через фиксированную точку пространства
(с).
3. ν – частота – число колебаний поля в секунду. Единица измерения – герц (Гц).
1 Гц = сек -1.
ν = 1/Р.
(4.7.23)
4. σ – волновое число – число волн, приходящихся на 1 см.
=1/λ.
(4.7.24)
5. Si – скорость распространения – скорость, с которой фронт волны движется через какую-либо среду.
Si = ν•λ (см/с);
(4.7.25)
Sвак ≈ 3•1010 (см/с).
6. Φ – мощность излучения – энергия потока, падающего на данную поверхность за 1 с.
Φ =ΔЕ/Δt (Вт). |
(4.7.26) |
794
I – интенсивность (мощность, приходящаяся на единицу телесного
угла).
Дискретные свойства электромагнитного излучения:
Е – энергия излучения (Дж или Ккал).
Е= h•ν.
(4.7.27)
1 кал ≈ 4,2 Дж.
h– постоянная Планка.
h = 6,63•10-27 эрг·с = 6,6•10-34 Дж·с.
Выражая ν через λ и σ, получим
Е= h•Si / λ = h•Si•σ.
(4.7.28)
4.7.2.1.2 Электромагнитный спектр
Электромагнитные волны классифицируются по длине волны или связанной с ней частотой волны. Совокупность длин волн или частот (энергий) представляет собой электромагнитный спектр излучения. Электромагнитный спектр охватывает огромную область длин волн или энергий. Качественную характеристику основных областей электромагнитного излучения изобразим в виде логарифмической шкалы (таблица 4.7.3).
Различные участки электромагнитного спектра отличаются по способу излучения и приема волн, принадлежащих тому или иному участку спектра. По этой причине между различными участками электромагнитного спектра нет резких границ, но каждый диапазон обусловлен своими особенностями.
Радиоволны изучает классическая электродинамика. Инфракрасное световое и ультрафиолетовое излучение изучает как классическая оптика, так
иквантовая физика. Рентгеновское и гамма-излучение изучается в квантовой
иядерной физике.
Рассмотрим спектр электромагнитных волн более подробно.
795
Таблица 4.7.3 – Шкала электромагнитных излучений
Радиоволны. Радиоволны представляют собой электромагнитные волны, длины которых более 1 мм (частота меньше 1011 Гц) и менее 10 км (частота выше 105 Гц).
Радиоволны делятся на:
-длинные волны в интервале длин от 10 км до 300 м (частота в диапазоне 105 Гц – 106 Гц);
-средние волны в интервале длин от 300 м до 100 м (частота в диапазоне 106 Гц – 3•106 Гц);
-короткие волны в интервале длин волн от 100 м до 10 м (частота в диапазоне 3•106 Гц – 3•107 Гц);
-ультракороткие волны с длиной волны меньше 10 м (частота больше
3•107 Гц).
Ультракороткие волны в свою очередь делятся: а) на метровые волны; б) сантиметровые волны; в) миллиметровые волны.
Волны с длиной волны меньше чем 1 м (частота больше, чем 3•108 Гц) называются микроволнами, или волнами сверхвысоких частот (СВЧ-волны).
Волны всех радиодиапазонов широко используются в технике – дециметровые и ультракороткие метровые волны применяются для телевещания и радиовещания в диапазоне ультракоротких волн, обеспечивая высокое качество приема сигнала в пределах зоны прямого распространения волн. Радиоволны метрового и километрового диапазона применяются для радиовещания и радиосвязи на больших расстояниях в пределах Земли благодаря отражению волн от ионосферы планеты. Впрочем, сегодня этот вид связи отходит в прошлое благодаря развитию спутниковой связи.
Сантиметровые волны, подобно дециметровым и метровым радиоволнам, практически не поглощаются атмосферой и поэтому ши-роко используются в спутниковой и сотовой связи и других телекоммуникационных системах.
Более короткие СВЧ-волны также находят применения в промышленности и в быту. Достаточно упомянуть про микроволновые печи, которыми сегодня оснащены и промышленные хлебопекарни, и домашние

796
кухни. Действие микроволновой печи основано на быстром вращении электронов в устройстве, которое называется клистрон. В результате электроны излучают электромагнитные СВЧ-волны определенной частоты, при которой они легко поглощаются молекулами воды. Когда помещают еду в микроволновую печь, молекулы воды, содержащиеся в еде, поглощают энергию микроволн, движутся быстрее и таким образом разогревают еду. Иными словами, в отличие от обычной духовки или печи, где еда разогревается снаружи, микроволновая печь разогревает ее изнутри.
Инфракрасное, видимое и ультрафиолетовое излучения.
Инфракрасное, световое, включая ультрафиолетовое, излучения составляют оптическую область спектра электромагнитных волн в широком смысле этого слова. Близость участков спектра перечисленных волн обусловила сходство методов и приборов, применяющихся для их исследования и практического применения. Исторически для этих целей применяли линзы, дифракционные решетки, призмы, диафрагмы, оптически активные вещества, входящие в состав различных оптических приборов (интерферометров, поляризаторов, модуляторов и пр.).
Оптический спектр занимает диапазон длин электромагнитных волн в интервале от 1 мкм до 10 нм. Верхняя граница оптического диапазона определяется длинноволновой границей инфракрасного диапазона, а нижняя
– коротковолновой границей ультрафиолетового.
Излучение оптического диапазона (видимый свет и ближнее инфракрасное излучение) свободно проходит сквозь атмосферу, может быть легко отражено и преломлено в оптических системах. Источники: тепловое излучение (в том числе Солнца), флуоресценция, химические реакции, светодиоды.
Лучи инфракрасной части спектра человек ощущает непосредственно кожей – как тепло. Если вы протягиваете руку в направлении огня или раскаленного предмета и чувствуете жар, исходящий от него, вы воспринимаете как жар именно инфракрасное излучение. У некоторых животных (например, у норных гадюк) есть даже органы чувств, позволяющие им определять местонахождение теплокровной жертвы по инфракрасному излучению ее тела.
Поскольку большинство объектов на поверхности Земли излучает энергию в инфракрасном диапазоне волн, детекторы инфракрасного излучения играют немаловажную роль в современных технологиях обнаружения. Инфракрасные окуляры приборов ночного видения позволяют людям «видеть в темноте», и с их помощью можно обнаружить не только людей, но и технику, и сооружения, нагревшиеся за день и отдающие ночью свое тепло в окружающую среду в виде инфракрасных лучей. Детекторы инфракрасных лучей широко используются спасательными службами, например, для обнаружения живых людей под завалами после землетрясений или иных стихийных бедствий и техногенных катастроф.
Свет представляет собой видимый участок спектра электромагнитных

797
волн, длины волн которых занимают интервал от 400 до 760 нм. Таким образом, область, воспринимаемая человеческим глазом, очень мала. Каждой спектральной составляющей оптического излучения может быть поставлен в соответствие определенный цвет. Окраска спектральных составляющих оптического излучения определяется их длиной волны. Цвет излучения изменяется по мере уменьшения его длины волны следующим образом: красный, оранжевый, желтый, зеленый, голубой, синий, фиолетовый.
Цвета видимого излучения, соответствующие монохроматическому излучению, называются спектральными. Спектр и спектральные цвета можно увидеть при прохождении узкого светового луча через призму или какуюлибо другую преломляющую среду.
Человеческий глаз представляет собой идеальный инструмент для регистрации и анализа электромагнитных волн этого диапазона. Это обусловлено двумя причинами. Во-первых, как отмечалось, волны видимой части спектра практически беспрепятственно распространяются в прозрачной для них атмосфере. Во-вторых, температура поверхности Солнца (около 5000°С) такова, что пик энергии солнечных лучей приходится именно на видимую часть спектра. Таким образом, наш главный источник энергии излучает огромное количество энергии именно в видимом световом диапазоне, а окружающая нас среда в значительной мере прозрачна для этого излучения.
К ультрафиолетовым лучам относят электромагнитное излучение с длиной волны от 10 до 400 нм. В этой части спектра излучение начинает оказывать влияние на жизнедеятельность живых организмов. Мягкие ультрафиолетовые лучи в солнечном спектре (с длинами волн, приближающимися к видимой части спектра), например, вызывают в умеренных дозах загар, а в избыточных – тяжелые ожоги. Жесткий (коротковолновый) ультрафиолет губителен для биологических клеток и поэтому используется, в частности, в медицине для стерилизации хирургических инструментов и медицинского оборудования, убивая все микроорганизмы на их поверхности.
Все живое на Земле защищено от губительного влияния жесткого ультрафиолетового излучения озоновым слоем земной атмосферы, поглощающим большую часть жестких ультрафиолетовых лучей. Если бы не этот естественный щит, жизнь на Земле едва ли бы вышла на сушу из вод Мирового океана. Однако, несмотря на защитный озоновый слой, какая-то часть жестких ультрафиолетовых лучей достигает поверхности Земли и способна вызвать рак кожи, особенно у людей, имеющих от рождения бледную кожу и плохо загорающих на солнце.
Рентгеновское и гамма-излучение. В области рентгеновского и гамма-излучения на первый план выступают квантовые свойства излучения.
Рентгеновское излучение возникает при торможении быстрых заряженных частиц (электронов, протонов и пр.), а также в результате процессов, происходящих внутри электронных оболочек атомов.
798
Гамма излучение является следствием явлений, происходящих внутри атомных ядер, а также в результате ядерных реакций. Граница между рентгеновским и гамма-излучением определяется условно по величине кванта энергии, соответствующего данной частоте излучения.
Рентгеновское излучение составляют электромагнитные волны с длиной от 10 нм до 10-3 нм, гамма излучение составляют электромагнитные волны с длиной волны меньше 10-3 нм.
Рентгеновские лучи проникают сквозь мягкие ткани организма и поэтому незаменимы в медицинской диагностике.
Самые короткие по длине волны и самые высокие по частоте и энергии лучи в электромагнитном спектре – это γ-лучи (гамма-лучи). Они состоят из фотонов сверхвысоких энергий и используются сегодня в онкологии для лечения раковых опухолей (а точнее, для умерщвления раковых клеток). Однако их влияние на живые клетки столь губительно, что при этом приходится соблюдать крайнюю осторожность, чтобы не причинить вреда окружающим здоровым тканям и органам.
В заключение важно еще раз подчеркнуть, что, хотя все описанные типы электромагнитного излучения проявляют себя внешне по-разному, по своей сути они являются достаточно близкими.
Все электромагнитные волны в любой части спектра представляют собой распространяющиеся в вакууме или среде поперечные колебания электрического и магнитного полей, все они распространяются в вакууме со скоростью света и отличаются друг от друга лишь длиной волны и, как следствие, энергией, которую они переносят.
Остается только добавить, что названные границы диапазонов носят достаточно условный характер. Например, микроволновые излучения с большими длинами волн нередко и справедливо относятся к сверхвысокочастотному диапазону радиоволн. Отсутствуют четкие границы и между жестким ультрафиолетовым и мягким рентгеновским, а также между жестким рентгеновским и мягким гамма-излучением.
4.7.2.1.3 Спектры атомов и их характеристики
Каждый атом имеет свой неповторимый набор энергетических состояний (рисунок 4.7.15). Переходы между этими состояниями приводят к возникновению спектра, который характерен для данного сорта атомов. Спектры атомов состоят из отдельно стоящих линий, поэтому они называются линейными, или дискретными. Каждая спектральная линия есть результат изменения состояния атома. Состояние атома, энергия которого минимальна, называется основным. Остальные состояния называются возбужденными. Время жизни атома в возбужденном состоянии составляет ≈
10ˉ с.

799
Рисунок 4.7.15 – Схема квантовых переходов: 1 – основной энергетический уровень; 2 – уровень излучения; 3 – уровень возбуждения
При переходе атома из возбужденного состояния в основное избыточная часть энергии излучается в виде кванта света. Такой процесс происходит самопроизвольно, так как атом переходит в состояние с меньшей энергией. Переход с основного состояния 1 в возбуженное 3 не может быть самопроизвольным, он может идти только вследствие внешнего воздействия. При этом квант света поглощается. Энергия атома увеличивается.
Любое вещество способно испускать или поглощать лучи определенной длины волны и определенной энергии. Эта энергия зависит от строения атома или молекулы вещества. В видимой и ультрафиолетовой областях спектра поглощение или испускание энергии связа-но с переходами электронов с одних орбиталей на другие (рисунок 4.7.16).
Рисунок 4.7.16 – Квантовые переходы и соответствующие им спектральные линии
В случае испускания атомами квантов света получаются спектры испускания (в спектре появляются светлые линии – спектральные максимумы). В случае поглощения квантов света – спектры поглощения (в спектре появляются темные линии поглощения – спектральные минимумы).
Спектральные линии, возникающие при переходе в основное состояние (спектры испускания) или из основного на возбужденное (спектры поглощения), называются резонансными. Если они расположены в УФили
800
видимой областях спектра, то спектр называется оптическим.
Все химические соединения взаимодействуют с электромагнитным излучением, уменьшая интенсивность потока излучения.
Методы, основанные на измерении уменьшения (ослабления) интенсивности излучения, прошедшего через анализируемое вещество, называются методами абсорбционной спектроскопии.
Различают:
-Молекулярный абсорбционный анализ – поглощение излучения молекулами или ионами.
-Атомно-абсорбционный – поглощение излучения возбужденными атомами.
4.7.2.1.4 Оптические методы анализа. Классификация
1.По изучаемым объектам различают: атомный и молекулярный спектральный анализ.
2.По характеру взаимодействия электромагнитного излучения с веществом:
- Атомно-абсорбционный анализ – измерение поглощения монохроматического излучения атомами определяемого вещества в газовой фазе после атомизации вещества.
- Молекулярный абсорбционный анализ – измерение светопоглощения молекулами или ионами изучаемого вещества.
- Эмиссионный спектральный анализ – измерение интенсивности света, излучаемого веществом (чаще всего – атомами или ионами) при его энергетическом возбуждении, например, в плазме электрического разряда.
- Эмиссионная фотометрия пламени – измерение интенсивности испускания видимых, УФ-, рентгеновских лучей атомами, возбужденными в пламени.
- Люминесцентный анализ – измерение интенсивности излучения веществом под воздействием различных видов возбуждения.
- Спектральный анализ с использованием эффекта комбинационного рассеяния света – измерение интенсивности излучения при явлении комбинационного рассеяния света.
- Нефелометрический анализ – измерение рассеивания света частицами дисперсной системы.
- Турбидиметрический анализ – измерение ослабления интенсивности излучения (поглощение) при его прохождении через дисперсную среду.
- Рефрактометрический анализ – измерение показателей преломления при прохождении света через границу раздела прозрачных сред.
- Поляриметрический анализ – измерение величины оптического вращения (угла вращения плоскости поляризации света) оптически активными веществами.
В аналитической химии используются и некоторые другие оптические
802
глощающий раствор с толщиной l, рассеивается, преломляется, но большая его часть поглощается. Из раствора выходит поток It, интенсивность которого меньше I0. При прохождении монохроматического излучения через раствор, содержащий поглощающее вещество, поток излучения ослабляется тем сильнее, чем больше энергии поглощают частицы этого вещества.
Понижение I зависит от концентрации вещества и длины пути, проходимого излучением (толщины слоя).
Эта зависимость выражается законом Бугера – Ламберта – Бера (основным законом светопоглощения).
Количество электромагнитного излучения, поглощенное раствором, пропорционально концентрации поглощающих частиц и толщине слоя
lg (It /Io) = - k •l •C, |
(4.7.29) |
Где, I0 – интенсивность падающего потока;
It – интенсивность потока, прошедшего через раствор; С – молярная концентрация раствора (моль/л);
l – толщина поглощающего слоя (см);
k – коэффициент пропорциональности (постоянная величина). Величина -lg(It/Io) называется абсорбцией, или оптической плотностью А. Тогда основной закон светопоглощения принимает форму
А= k • l • C.
Если концентрация раствора выражена в моль/л, а толщина поглощающего слоя в см, то коэффициент k = ε, и основной закон светопоглощения принимает вид
А = ε• l • C, |
(4.7.30) |
где ε – молярный коэффициент поглощения.
Молярный коэффициент поглощения является мерой чувствительности фотометрических методов. Чем больше ε, тем выше чувствительность метода, тем меньшую концентрацию вещества можно определить.
Физический смысл ε: при C = 1 моль/л и толщине слоя l = 1 см ε = А. Молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине слоя 1 см.
Закон Бугера – Ламберта – Бера применим к растворам, содержащим несколько поглощающих веществ при условии, что между этими соединениями отсутствуют взаимодействия (закон аддитивности). Каждое из

803
веществ будет давать свой аддитивный вклад в экспериментально определяемую оптическую плотность:
Аобщ = А1 + А2 +А3 + … = ε1lC1 + ε2lC2 + ε3lC3 + … . (4.7.31)
Отношение интенсивности прошедшего излучения к интенсивности падающего излучения называется пропусканием Т (прозрачностью). Это часть падающего излучения, прошедшего через раствор (обычно выражается в %).
(4.7.32)
Оптическая плотность и пропускание связаны между собой соотношением
А= -lgT . (4.7.33)
Закон Бугера – Ламберта – Бера непосредственно в химическом анализе не применяют, так как невозможно измерить I0 и It. Кроме того, возможно взаимодействие между излучением и стенками кюветы (поглощение, излучение и др.), излучение ослабевает и вследствие взаимодействия с крупными молекулами (например, растворителя). Чтобы компенсировать эти потери, сравнивают интенсивность прошедшего через раствор потока с интенсивностью потока, прошедшего через раствор холостого опыта, и измеряют оптическую плотность, близкую к истинной.
Ограничения и условия применимости закона Бугера – Ламберта – Бера. Закон Бугера – Ламберта – Бера отражает линейную зависимость оптической плотности А от концентрации С при постоянной толщине поглощающего слоя l (закон Бера) и, наоборот, зависимость А от l при постоянной С (закон Бугера – Ламберта).
Во втором случае зависимость является правилом, из которого нет исключений.
Зависимость А от С при постоянном значении l в идеале должна носить линейный характер (рисунок 4.7.18).

804
Рисунок 4.7.18 – Зависимость оптической плотности вещества А от концентрации С при соблюдении основного закона светопоглощения
При выполнении закона Бера график зависимости оптической плотности от концентрации представляет собой прямую, проходящую через начало координат (рисунок 4.7.18), а функция А = f(λ), графическая зависимость которой называется спектром поглощения, имеет один и тот же вид, независимо от толщины слоя и концентрации раствора, и положение максимума поглощения сохраняется (рисунок 4.7.19).
Рисунок 4.7.19– Спектр поглощения одного и того же вещества при соблюдении закона Бугера – Ламберта – Бера. С1 < С2 < С3 < С4

805
Рисунок 4.7.20 – Зависимость оптической плотности вещества А от концентрации С при соблюдении основного закона светопоглощения 1, положительных 2 и отрицательных 3 отклонениях от закона
Однако линейность зависимости А от С соблюдается не всегда
(рисунок 4.7.20).
Отклонения от закона бывают истинные, т.е. носящие фундаментальный характер, химические и инструментальные.
Истинные ограничения применимости закона Бугера – Ламберта –
Бера:
1.Закон Бера справедлив для разбавленных растворов. При вы-соких концентрациях (С > 0,01 М) среднее расстояние между части-цами поглощающего вещества уменьшается до такой степени, что каждая частица влияет на распределение заряда соседних частиц, что, в свою очередь, может изменить способность частиц поглощать излучение данной длины волны.
2.Коэффициент ε в уравнении (4.7.8) зависит от показателя преломления среды η. Если С < 0,01 М, то η остается почти таким же, каким был у чистого растворителя, отклонений не наблюдается. Увеличение концентрации раствора приводит к значительному изменению показателя преломления η и отклонению от закона Бера (показатели преломления разбавленных растворов и растворителя отличаются несущественно).
Другие причины отклонения от закона Бугера – Ламберта – Бера связаны со способом измерения А или с химическими изменениями, возникающими вследствие изменения концентрации. Поэтому они называются химическими и инструментальными.
Химические (кажущиеся) ограничения применимости закона Бугера – Ламберта – Бера: Уравнение (7.8) соблюдается для систем, в которых светопоглощающими центрами являются частицы одного сорта, т.е. отсутствует химическое взаимодействие. Если при изменении концентрации будет меняться природа этих частиц, вследствие, например, кислотноосновного взаимодействия, полимеризации, диссоциации, комплексообразования и т. д., то зависимость А = f(С) не будет линейной, так как молярный коэффициент поглощения вновь образующихся и исходных частиц не будет одинаковым. Классическим примером является раствор бихромата

806
калия, в котором при разбавлении устанавливается следующее равновесие:
Cr2O72- + H2O ↔ 2 HСrO4- ↔ 2 H+ + 2 CrO42-.
Молярные коэффициенты бихромата, гидрохромата и хромата довольно сильно различаются. Зависимость оптической плотности от общей концентрации хрома не будет линейной.
Эти отклонения являются кажущимися, так как они возникают в результате смещения химического равновесия. Их можно предсказать, изучить и устранить (создать нужное значение рН, подобрать растворитель и т.д.).
Инструментальные ограничения применимости закона Бугера – Ламберта – Бера:
1. Закон справедлив для монохроматического излучения. Строго говоря, уравнение (4.7.8) следует записывать в виде
Аλ = ελ · l · C. (4.7.34)
Индекс λ указывает, что величины А и ε относятся к монохроматическому свету с длиной волны λ. Немонохроматичность светового потока связана с несовершенством оптических приборов. Отклонение от закона Бера менее заметно, если длина волны не приходится на часть спектра с резким изменением оптической плотности. На практике измерение А стремятся проводить в максимуме светопоглощения.
2.Температура при измерениях должна оставаться постоянной хотя бы
впределах нескольких градусов.
3.Пучок света должен быть параллельным.
4.Измеряемая оптическая плотность должна находиться в интервале от 0,2 до 0,8, так как в этом случае погрешность измерения мини-мальна (теоретически А может находиться в интервале от 0 до ∞).
4.7.2.2.2 Аппаратурное оформление метода
Независимо от области спектра приборы для измерения пропускания или поглощения излучения состоят из пяти основных узлов (рисунок 4.7.21).
Примечание: 1 – источник излучения; 2 – монохроматор (устройство, выделяющее определенную λ); 3 – прозрачные кюветы с исследуемым раствором и растворителем (раствором сравнения); 4 – приемник излучения (детектор), который превращает энергию излучения в измеряемый сигнал; 5 – измерительное или регистрирующее устройство (индикатор сигнала)
Рисунок 4.7.21 – Блок-схема приборов для измерения поглощения излучения

807
Индикатор часто снабжают lg шкалой, непосредственно указывающей на оптическую плотность. Обычно сначала измеряют оптическую плотность холостого раствора (раствора сравнения), затем настраивают индикатор на 0, после этого измеряют оптическую плотность исследуемого раствора.
Для измерения поглощения в видимой области используют колориметры, фотоэлектроколориметры (ФЭК), спектрофотометры (СФ). В ультрафиолетовой (УФ-) и инфракрасной (ИК-) областях из-мерения осуществляют только на спектрофотометрах.
Колориметры. Детектором служит глаз человека. Однако мы можем лишь сравнивать окраску и неспособны дать численную информацию об оптической плотности. Поэтому в колориметрическом методе используются эталоны для сравнения их окраски с окраской анализируемых растворов.
Для этого проводят:
1. Сравнение пробы с серией эталонов.
Часто применяют колориметрические пробирки Несслера, которые прокалиброваны таким образом, что толщина слоя всех растворов одинакова. Источник излучения – дневной свет.
2. Сравнение неизвестного раствора с одним эталонным раствором. Оба раствора помещают в колориметрические пробирки и меняют толщину слоя одного из них до уравнивания интенсивности. При этом:
Ах = А1,
· ε• lx •Cх = ε •l1 • C1.
После этого вычисляют концентрацию анализируемого раствора Сх:
(4.7.35)
Этот способ положен в основу колориметра Дюбоска. Недостатки:
1.Всегда необходим эталон или серия эталонов.
2.Интенсивность невозможно сравнивать в присутствии других окрашенных веществ.
3.Глаз человека не столь чувствительный по сравнению с при-борами. Поэтому эти методы лишь приблизительные.
Фотометры и спектрофотометры.
Фотометры (фотоэлектроколориметры) – это простые, недорогие
приборы.
Достоинства – удобство, простота в устройстве и обращении. Недостатки – нет строгой монохроматизации излучения.
Однако, если метод не требует монохроматизации (чаще широкий спектр), анализы можно проводить с высокой точностью. Спектрофотометры

808
– приборы, обеспечивающие более точное измерение вследствие монохроматизации. Монохроматизаторами служат призма или дифракционная решетка, позволяющие менять длину волны.
Сравнительные характеристики фотометров и спектрофотометров представлены в таблице 4.7.4.
Таблица 4.7.4 – Сравнительные характеристики фотометров и спектрофотометров
Характеристика |
Фотометры |
|
|
Спектрофотометры |
|
||||
|
|
|
|
|
|||||
Источник |
|
Нить |
вольфрамовой |
Вольфрамовые лампы λ от 320 (бл. УФ-) |
|||||
излучения |
|
лампы. |
|
|
|
до 2500 (ИК-). |
|
|
|
|
|
Используют в интервале λ |
Для УФ-излучения – водородная и |
||||||
|
|
от 320 до 1000 нм |
|
дейтериевая лампы. |
|
|
|||
|
|
|
|
|
|
Для ИК-излучения – твердое тело, |
|||
|
|
|
|
|
|
нагретое электрическим током |
|
||
Монохроматор |
Светофильтры. |
|
|
1. Призмы (преломляют излучение, после |
|||||
ы (устройства, |
Поглощают |
большую |
прохода призмы. Излучение выходит в |
||||||
разлагающие |
|
часть |
спектра |
и |
виде |
узкой линии), для УФ – кварцевые, |
|||
излучение |
на |
пропускают |
|
|
для видимого – стеклянные. |
|
|||
разные λ) |
|
ограниченный участок λ |
|
2. Дифракционные решетки (кусок стекла |
|||||
|
|
|
|
|
|
или другого прозрачного материала, на |
|||
|
|
|
|
|
|
который нанесена серия штрихов; после |
|||
|
|
|
|
|
|
освещения решетки |
каждый |
штрих |
|
|
|
|
|
|
|
становится новым источником излучения; |
|||
|
|
|
|
|
|
в результате излучение разлагается на |
|||
|
|
|
|
|
|
разные λ). |
|
|
|
|
|
|
|
|
|
3. Двойные монохроматоры (состоят из 2 |
|||
|
|
|
|
|
|
призм или решеток) |
|
|
|
Кюветы |
|
Стеклянные, |
иногда |
Стеклянные или кварцевые (для УФ) |
|||||
|
|
пластиковые с l от 0,1 до |
|
|
|
|
|||
|
|
10 см |
|
|
|
|
|
|
|
Детекторы |
|
Фотоэлементы |
или (при |
Фотоэлементы или фотоумножители. В |
|||||
|
|
измерении |
излучения |
с |
УФ – чаще фоумножители, они более |
||||
|
|
низкой интенсивностью) |
|
чувствительны. В ИК-термопара или |
|||||
|
|
|
|
|
|
болометр |
|
|
|
Индикаторы |
|
Обычно |
гальванометры, |
Чаще |
вольтметры |
или |
другие |
||
|
отградуированные на λ |
|
электроприборы |
|
|
||||
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
809
4.7.2.2.3 Фотометрический метод анализа
Фотометрический метод используется для определения ионов в растворах, поглощающих электромагнитное излучение в УФ-, видимой и ближней ИК-областях спектра.
Фотометрическое определение состоит из двух частей:
1.Переведение определяемого компонента в соединение, поглощающее электромагнитное излучение.
2.Измерение интенсивности поглощения раствором полученно-го соединения.
Фотометрические методы разработаны практически для всех элементов. Однако не для всех ионов существуют реакции получения соединений, растворы которых поглощают в УФ-, видимой и ближней ИКобластях спектра. Поэтому существуют прямые и косвенные методы фотометрического определения.
1.Прямой:
X+R=XR,
Где, Х – определяемый ион;
ХR – соединение, поглощающее электромагнитное излучение. Определяемый компонент переводят в соединение, поглощающее электромагнитное излучение.
2. Косвенный:
MR+X=MX,
Где, MR – соединение, поглощающее электромагнитное излучение;
Х– определяемый ион.
Копределяемому компоненту добавляют соединение, поглощающее электромагнитное излучение.
3. Косвенный:
Х +R=XR,
Где, Х – определяемый ион;
ХR – нерастворимое соединение.
При взаимодействии реагента и определяемого иона образуется осадок, который отделяют, переводят в раствор и один из компонентов определяют фотометрическим методом.
К косвенным методам также относят фотометрическое титрование. Это разновидность титриметрического анализа, при котором точку эквивалентности определяют фотометрически.
Области применения фотометрического анализа:
1.Для определения многих катионов и анионов.
2.В химической, металлургической и других отраслях промышленности.
810
3.Для анализа руд, минералов и других природных объектов.
4.В сельском хозяйстве.
5.В медицине и биологии.
6.В аналитическом контроле загрязнений окружающей среды и решении экологических проблем.
8.Для контроля производства, определения примесей и решения других вопросов в заводской и научно-исследовательской лабораториях.
9.Для исследования различных реакций, установления состава и устойчивости образующихся соединений, для изучения равновесий в растворах и определения аналитических констант.
Отличительные особенности и достоинства метода:
1.Относительно прост в выполнении.
2.Недорогое и доступное оборудование.
3.Используется для определения практически всех ионов (элементов).
4.Используется для определения больших и малых концентраций.
5.Высокая чувствительность метода (низкий предел обнаружения). Чувствительность Н (Н = tg α) – тангенс угла наклона градуировочного
графика, который является графиком зависимости величины физического параметра от концентрации определяемого элемента для серии стандартных образцов с известным содержанием этого элемента.
Предел обнаружения – наименьшая концентрация, которая может быть обнаружена с разумной достоверностью, т.е. та концентрация, которой соответствует сигнал, равный утроенному стандартному отклонению результатов измеряемого фона.
6.Высокая точность. Погрешность фотометрических методов составляет не более 2–5 %.
7.Высокая избирательность многих фотометрических реакций, т.е. возможность определения элементов в сложных пробах без предварительного разделения компонентов.
Выбор оптимальных условий проведения фотометрических определений. Для проведения фотометрической реакции определяемый компонент переводят в соединение, обладающее значительным поглощением. Чаще всего его связывают в комплексное соединение.
При использовании фотометрического титрования применяют те же реакции, что и в обычных методах, однако о содержании вещества судят не по интенсивности поглощения, а по количеству затраченного титранта.
Условия проведения фотометрических реакций должны быть предварительно тщательно изучены.
Предварительное изучение рабочих условий включает:
1.Выбор длины волны.
В спектрофотометрах рекомендуется проводить измерения при длине волны λ, соответствующей максимальному значению А (λmax). Для этого строится зависимость А от λ (снимается спектр поглощения) по всей длине шкалы, для работы выбирается длина волны, при которой поглощение
812
раствора достаточно интенсивная, пользуются кюветами менее 1 см. Практически поступают следующим образом: в кювету наливают раствор средней концентрации из эталонного ряда и измеряют оптическую плотность, которая должна находиться в интервале от 0,4 до 0,6 (в этом случае погрешность измерения минимальна). Если А больше этих значений, то следует взять кювету с меньшим значением l. Если А меньше, чем 0,4–0,6, то нужно использовать кювету большей толщины. На практике чаще всего используются кюветы толщиной 1 см.
5. Выбор раствора сравнения.
В качестве раствора сравнения в зависимости от условий чаще всего используют:
–растворитель;
–раствор реагента, если он сам поглощает излучение;
–раствор холостого опыта (содержит все компоненты, кроме определяемого);
–раствор анализируемого объекта.
6.Построение градуировочного графика, отражающего зависимость оптической плотности от концентрации компонента (А от С).
Готовят серию эталонных (стандартных) растворов с различными концентрациями определяемого компонента. В выбранных условиях (λ, l и т.д.) измеряют оптическую плотность каждого раствора и строят градуировочный график. Определяют интервал соблюдения закона Бугера – Ламберта – Бера (прямолинейность графика).
7.Расчет пределов обнаружения и определения минимальной и максимальной концентрации данного компонента. Это надежно устанавливается лишь с использованием методов математической статистики.
Методы определения концентрации окрашенных растворов:
1.Метод градуировочного графика. Записывают спектр поглощения раствора вещества и находят длину волны, соответствующую максимуму поглощения. Затем готовят серию стандартных растворов с различным содержанием определяемого компонента и измеряют их оптическую плотность при выбранной длине волны и толщине слоя. Необходимо, чтобы выбранный интервал концентрации соответствовал области возможных изменений концентраций анализируемых растворов. Строят градуировочный график в координатах А – C. В случае подчинения закону Бугера – Ламберта – Бера и при измерении оптической плотности относительно растворителя или холостого опыта, график представляет собой прямую (см. рисунок 7.5), проходящую через начало координат. Затем проводят ту же фотометрическую реакцию с раствором неизвестной концентрации определяемого компонента. Измеряют оптическую плотность исследуемого раствора Ах и по графику находят концен-трацию Сх вещества в растворе. Определив оптическую плотность исследуемого раствора Ах, находят ее значение на оси координат, проводят перпендикуляр до пересечения с

813
графиком, а затем перпендикуляр – на ось абсцисс (С). Точка пересечения с осью – это и есть значение концентрации Сх.
Для интенсивно окрашенных растворов (А ≥ 1) используется метод дифференциальной фотометрии. Дифференциальный метод применяют для повышения воспроизводимости результатов анализа при определении больших количеств веществ, когда нарушается основной закон светопоглощения или когда значения оптических плотностей выходят за пределы шкалы прибора, а дальнейшее разбавление раствора может привести к увеличению погрешности определения.
Вкачестве раствора сравнения используют растворы с меньшей или большей концентрацией определяемого компонента, чем в исследуемом растворе. Рассчитать концентрацию вещества в методе дифференциальной фотометрии можно также с использованием градуировочного графика (рисунок 4.7.5, 4.7.9). При построении градуировочных графиков в дифференциальной фотометрии различают два варианта: метод односторонней дифференциальной фотометрии и метод двухсторонней дифференциальной фотометрии. В методе односторонней дифференциальной фотометрии для построения градуировочного графика используют раствор сравнения с концентрацией меньшей, чем концентрации стандартных растворов, т. е. С0 < Cср. В методе двухсторонней дифференциальной фотометрии используют стандартные растворы с концентрацией большей и меньшей, чем концентрация раствора сравнения (рисунок 4.7.22)
Рисунок 4.7.22 – Градуировочный график для определения концентрации компонента растворе методом двусторонней дифференциальной фотометрии
2. Алгебраический метод (метод молярного коэффициента поглощения). Метод используется только, если известно, что растворы подчиняются закону Бера (прямолинейная зависимость А от С). Тогда готовят два раствора: эталонный Сэ и испытываемый Сх. Для каждого из них справедливы выражения:
Аэ = ε •l • Cэ, |
Ах = ε• l • Cх. |
Так как ε и l одинаковы, то,

814
(4.7.36)
Если ε и l заранее известны, то Cх можно рассчитать сразу из формулы
(4.7.37)
3. Метод добавок. Этот метод используется при анализе растворов сложного состава, при этом учитывается влияние посторонних компонентов.
Вэтом методе сначала измеряют оптическую плотность Ах анализируемого раствора, содержащего анализируемый компонент неиз-вестной концентрации Cх. Затем в анализируемый раствор вводят из-вестное количество определяемого компонента Сст и измеряют Ах+ст. Оптическая плотность анализируемого раствора
Ах = ε •·l • Cх.
Оптическая плотность анализируемого раствора с добавкой
Ах+ст = ε• l • (Cх + Сст).
(4.7.38)
Отсюда
Разделив левую и правую части уравнения на Сх, получим:

815
Отсюда
(4.7.39)
Графический вариант: изучают зависимость Ах+ст от Сст. Если построить график этой зависимости при различных известных концентрациях добавленного компонента, то экстраполяция этой прямой до пересечения с осью абсцисс даст отрезок, равный Сх (рисунок 4.7.23).
Рисунок 4.7.23 – Определение концентрации компонента методом добавок
Метод добавок обычно применяют для устранения мешающего действия посторонних примесей, а также в ряде случаев для оценки правильности методики определений. Этот метод позволяет создать одинаковые условия для фотометрирования исследуемого раствора и раствора с добавкой, поэтому его целесообразно применять для определения небольших количеств различных соединений в присутствии больших количеств посторонних веществ. Метод добавок требует обязательного соблюдения основного закона светопоглощения.
4.7.2.2.4 Фотометрическое титрование
Фотометрическое титрование – косвенный метод анализа. В этом методе используют те же реакции, что и в обычных методах, однако о содержании вещества судят не по интенсивности поглощения, по количеству затраченного титранта. Это разновидность титриметрического анализа, при котором точку эквивалентности определяют фотометрически.
Применяют в случаях, если:
-в результате титрования образуется окрашенное соединение;
-при титровании веществ, поглощающих в УФ- и видимых областях спектра;
-при титровании окрашенных растворов;
-цвет индикатора изменяется постоянно;
-при титровании разбавленных растворов.
816
Необходимое условие – не использовать избыток реагента. Необходимо, чтобы при эквивалентном количестве реагента было достигнуто практически полное связывание определяемого компонента.
В отсутствие индикатора необходимо, чтобы титруемые растворы, титрант или продукт реакции имели собственную полосу поглощения.
Спектрофотометрическое и фотометрическое титрование заключается в том, что в процессе титрования исследуемого раствора фиксируют изменение оптической плотности. Точку эквивалентности устанавливают по максимальному изменению оптической плотности. Предполагается, что поглощение раствора подчиняется закону Бугера – Ламберта – Бера. Титрование проводят непосредственно в спектрофотометрах, снабженных специальными кюветными крышками с отверстиями для ввода кончика полумикробюретки и мешалки, в кюветах объемом 25 мл.
Градуировочные графики строят при λmax в координатах А – Vтитранта. Точка пересечения двух прямых (точка перегиба) соответствует точке эквивалентности. Расчеты проводят как при обычном титровании по формуле
C ∙• V = С ∙• V , (4.7.40)
где C – молярная концентрация эквивалента (нормальность) определяемого компонента, моль/л;
V – объем раствора, взятый для анализа, мл;
С – молярная концентрация эквивалента (нормальность) рабочего раствора, моль/л;
V – объем рабочего раствора, пошедший на титрование, мл. Кривые фотометрического титрования могут иметь различный вид (рисунок 4.7.11).
Точность установления точки эквивалентности тем больше, чем резче выражен излом на кривой вблизи этой точки. Это наблюдается для практически необратимых реакций и для реакций, с большой константой равновесия, например, при образовании устойчивых комплексных соединений. В тех случаях, когда на кривых спектрофотометрического титрования отсутствует резкий излом, точку эквивалентности находят, экстраполируя касательные к участкам кривой титрования (рисунок 4.7.24).

817
Рисунок 4.7.24 – Различные формы кривых фотометрического титрования: а –
определяемый компонент и продукт реакции не поглощают излучение, титрант – поглощает; б – титруемый раствор и титрант не поглощают, продукт – поглощает; в – поглощает определяемый компонент, продукт и титрант – не поглощают; г – определяемый компонент и тирант поглощают, продукт – не поглощает; д, е – продукт и титрант поглощают, определяемый компонент – нет, при этом: д – продукт поглощает слабее, чем титрант; е – продукт поглощает сильнее, чем титрант
Точность спектрофотометрического титрования составляет ± 0,5% и превышает точность прямой спектрофотометрии.
4.7.2.3 Использование спектров атомов в аналитических целях
Мы уже знаем, что каждый атом имеет свой характерный спектр.
Имея спектр неизвестного сорта атомов и сравнивая его со спектрами известных атомов, можно идентифицировать неизвестные атомы, т. е. решить задачу качественного анализа.
Измеряя интенсивность линий в спектре исследуемого сорта атомов, можно рассчитать число атомов, участвующих в образовании спектра поглощения или испускания, т. е. решить задачу количественного анализа.
Однако обычно химики имеют дело не со свободными атомами, а с веществами. Атомы, входящие в состав веществ, находятся в сильном взаимодействии друг с другом, поэтому они теряют свои индивидуальные характеристики, связанные с внешними электронами. Следовательно, оптические спектры веществ не будут похожи на оптические спектры свободных атомов.
Для получения оптических спектров атомов необходимо создать такие условия, при которых вещество будет распадаться на свободные атомы.
Одним из простейших способов атомизации вещества является термический нагрев при введении его в пламя. При высокой температуре жидкие и твердые вещества будут переходить в газообразное состояние, а молекулы – диссоциировать на свободные атомы.
Однако не все атомы, входящие в состав исследуемого вещества, будут


820
разработан в 1955 г. австралийским исследователем А. Уолшем. Это метод количественного элементного анализа по атомным спектрам поглощения (абсорбции). Через слой атомных паров пробы, получаемых с помощью атомизатора, пропускают излучение в диапазоне 190–850 нм. В результате поглощения квантов света атомы переходят в возбужденные энергетические состояния. Этим переходам в атомных спектрах соответствуют так называемые резонансные линии, характерные для данного элемента.
Атомно-абсорбционный метод анализа заключается в измерении поглощения резонансного излучения атомами определяемого элемента.
Приборы для атомно-абсорбционного анализа – атомно-абсорбционные спектрометры – высокоавтоматизированные устройства, обеспечивающие воспроизводимость условий измерений, автоматическое введение проб и регистрацию результатов измерения.
В качестве примера на рисунке 4.7.25 приведена схема одного из спектрометров.
Источником линейчатого излучения в спектрометрах чаще всего служат одноэлементные лампы с полым катодом, заполняемые неоном. Перевод анализируемого объекта в атомизированное состояние и формирование поглощающего слоя пара определенной и воспроизводимой формы осуществляется в атомизаторе – обычно в пламени или трубчатой печи. Наиболее часто используют пламя смесей ацетилена с воздухом и ацетилена с N2O.
Рисунок 4.7.25 – Принципиальная схема пламенного атомно-абсорбционного спектрометра: 1 – источник излучения; 2 – пламя; 3 – монохроматор; 4 – фотоумножитель; 5 – регистрирующий или показывающий прибор
Прошедшее через пламя излучение попадает на входную щель монохроматора, установленную таким образом, что из спектра выделяется резонансная линия определяемого элемента, интенсивность которой измеряют фотоэлектрическим способом.
Уменьшение интенсивности резонансной линии вследствие поглощения атомами определяемого элемента, находящегося в пламени, измеряют, принимая интенсивность неослабленной линии за 100 %.
Величина поглощения резонансного излучения пропорциональна числу атомов, находящихся в поглощающем слое. Зависимость между ослаблением интенсивности излучения источника света и концентрацией вещества подчиняется закону Бугера – Ламберта – Бера:
821
(4.7.41)
Где, I0 – интенсивность резонансного излучения;
I – интенсивность излучения, прошедшего через поглощающий слой; k – коэффициент поглощения света в центре линии поглощения;
l – толщина поглощающего слоя.
Любой элемент можно определить с помощью атомно-абсорбционного метода, если его резонансная линия находится в области спектра, в которой работает прибор, и если элемент может быть атомизирован с помощью имеющихся в распоряжении методов. Трудно определяются элементы, соединения которых не полностью диссоциируют при температуре пламени, или те, которые образуют в пламени термостойкие соединения. В этом случае для атомизации образца и для повышения чувствительности анализа используют пламя с более высокой температурой и другие способы атомизации проб, например, графитовую кювету, лазер и др.
Достоинства атомно-абсорбционного анализа:
1.Метод характеризуется высокой чувствительностью, позволяющей определять некоторые элементы в концентрации 0,1– 0,005 мкг/мл и ниже.
2.Точность метода составляет 1–4 % в зависимости от величины определяемой концентрации.
3.Метод отличается быстротой и простотой выполнения, доступностью и несложностью применяемой аппаратуры.
4.Метод используется для определения большинства химических элементов.
5.Атомно-абсорбционный анализ используется для определения элементов в разных объектах: в сплавах, в чистых металлах, нефтепродуктах,
вреактивах, почвах, биологических жидкостях, водах и т.д.
6.Методы атомно-абсорбционного анализа применяют также для измерения некоторых физических и физико-химических величин – коэффициентов диффузии атомов в газах, температуры газовой среды, теплоты испарения элементов, для изучения спектров молекул, ис-следования процессов, связанных с испарением и диссоциацией соединений.
Ограничения метода – невозможность одновременного определения нескольких элементов при использовании линейчатых источников излучения и, как правило, необходимость переведения проб в раствор.
4.7.2.5 Эмиссионная фотометрия пламени
Метод эмиссионной фотометрии пламени является одним из вариантов эмиссионного спектрального анализа, в основе которого лежит использование спектров испускания атомов или молекул (эмиссионных спектров).
Метод основан на измерении интенсивности света, излучаемого
822
возбужденными атомами (или молекулами) при введении вещества в пламя горелки.
Аппаратура метода эмиссионной фотометрии пламени. Для проведения анализа используют пламенные фотометры, которые предназначены в основном для определения щелочных и щелочноземельных металлов в растворах, питьевых, минеральных, сточных, водах, винах, напитках, биологических жидкостях, фармацевтических препаратах, почвах, минералах. Поскольку спектры эмиссии атомов значительно проще молекулярных, то именно методы, основанные на их получении, стали широко применяться для массового многоэлементарного экспресс-анализа. Принципиальная схема приборов мало отличается от схемы, приведенной на рисунке 4.7.12.
Принцип метода заключается в следующем. Раствор распыляют в виде аэрозоля в пламя горелки, где происходит ряд сложных процессов, в результате которых образуются возбужденные атомы или молекулы. За счет энергии пламени легко возбуждаемым атомам вещества (чаще всего K, Na, Ca) сообщается избыточная энергия. Атомы этих металлов переходят в возбужденное состояние, характеризующееся переходом валентных электронов на более высокие энергетические уровни. Через 10-8 с происходит их возврат на основные уровни, что сопровождается выделением порций энергии (квантов света). Совокупность квантов света приводит к образованию светового потока с длиной волны, характерной для данных атомов.
Возникающее излучение определяемого элемента отделяется от постороннего с помощью монохроматора или светофильтра и, попадая на фотоэлемент, вызывает фототок, который измеряется с помощью гальванометра.
Качественный анализ проводят путем идентификации спектральных линий в спектре пробы, т.е. установления их длины волны, интенсивности и принадлежности тому или иному элементу.
Для расшифровки спектра и определения длины волны анализируемой линии пользуются спектрами сравнения, в которых длины волн отдельных линий указаны. Чаще всего для этой цели используют хорошо изученный спектр железа, имеющий характерные группы линий. Устанавливают длины волн спектральных линий. После этого идентифицируют линии в спектре пробы с помощью специальных таблиц, в которых указана принадлежность всех возможных спектральных линий определенным элементам (с указанием их числа, цвета, длины волны, потенциала ионизации). Считается, что элемент присутствует в пробе, если идентифицированы три или четыре его спектральные линии.
Градуировочные графики строят в координатах: сила фототока (I, ma) – концентрация определяемого вещества (С, мкг/мл) для постоянного режима работы фотометра. Графики строят по результатам анализа серии стандартных растворов.
823
Линейную зависимость I от С нарушают самопоглощение света невозбужденными атомами в области больших концентраций, ионизация в области малых концентраций, образование газообразных трудно диссоциирующих соединений.
Методы определения концентрации вещества в растворе.
Зависимость интенсивности излучения от концентрации атомов в пламени для резонансных линий имеет линейный характер лишь на небольшом участке концентраций. Для большей точности желательно работать в прямолинейном участке градуировочного графика, который может оказаться разным при работе на различных приборах. Поэтому независимо от выбранного способа определения неизвестной концентрации вещества в растворе нужно провести градуировку прибора. Для этого с помощью эталонных растворов необходимо найти зависимость между показаниями прибора и концентрацией соли металла в растворе.
Для определения концентрации вещества в растворе используют известные в аналитической химии методы, из которых наиболее часто:
–метод градуировочного графика;
–метод добавок;
–метод ограничивающих растворов.
Достоинства метода:
1. Экспрессность.
2. Точность метода составляет 2–4 %.
3. Чувствительность зависит от свойств аналитической линии, состава
пробы, аппаратуры. Предел обнаружения обычно находятся в интервале от
0,01 до 0,001 мкг/мл.
4.Более половины элементов периодической системы можно определить этим методом. Применение косвенных методов позволяет расширить число определяемых элементов. Например, P или Al определяют по гашению излучения Са.
5.Особую ценность метод приобретает при определении примесей щелочных и щелочноземельных элементов в самых разнообразных объектах.
6.Широкое распространение метод получил в геологии, биологии, медицине, агрохимии, металлургии, промышленности.
В заключение необходимо отметить, что в литературе нет едирообразия и четкости в терминологии этого метода: метод эмиссионной фотометрии пламени часто называют методом пламенной фотометрии или просто фотометрии пламени.
4.7.2.6 Люминесцентный метод анализа
Люминесценцией называют свойство веществ излучать свет под воздействием различных возбуждающих факторов.
Согласно определению С.И. Вавилова, люминесценцией называют излучение, избыточное над температурным (тепловым) и обладающее длительностью не менее чем 10-10 с, что превышает период световых

825
отражены процессы люминесценции):
1.Свечение дискретных центров – свечение возникает в тех случаях, когда поглощающими и излучающими центрами являются одни и те же частицы (атомы, ионы, молекулы). Этот механизм присущ веществам в газообразном состоянии, органическим и неорганическим веществам в растворах и чистым органическим веществам.
2.Рекомбинационное свечение – наблюдается в тех случаях, когда акты поглощения и излучения разделены не только по времени, но в пространстве (свечение кристаллофосфоров – сложных кристаллических веществ с дефектной структурой). Рекомбинационное свечение особенно характерно для кристаллов с ионной или ионно-ковалентной решеткой. Отделившиеся от центров или от некоторых мест решетки электроны имеют возможность быстро перемещаться по кристаллу вследствие того, что в нем существуют зоны свободного перемещения электронов, так называемые зоны проводимости. Движение электронов в этих зонах при не слишком низких температурах происходит с большой скоростью – много километров в секунду. Вследствие этого электрон рекомбинирует не с тем центром свечения, от которого он отделился, а с другим, также потерявшим электрон.
Свечения дискретных центров и рекомбинационное могут быть охарактеризованы по крайней мере четырьмя свойствами:
1) спектрами излучения, возбуждения и поглощения;
2) выходом;
3) поляризацией;
4) длительностью.
Природа люминесцентного излучения:
Происхождение люминесцентного излучения в простейшем виде поясняется схемой, представленной на рисунке 4.7.26.
Рисунок 4.7.26 – Энергетическая модель люминесценции:
S0 – основной электронный уровень (с колебательными подуровнями);
S1 и Т1 – возбужденные электронные уровни (синглетный и триплетный соответственно); прямыми вертикальными стрелками обозначены: поглощение (1), излучательные переходы: флуоресценция (2) и фосфоресценция (3); горизон-тальными стрелками – безызлучательные переходы: интеркомбинационная конверсия (4) и внутренняя конверсия (5)
826
При поглощении света происходит переход электронов с основного уровня S0 на колебательные подуровни возбужденного уровня (S1), соответствующего синглетному состоянию (антипараллельные спины). Затем за время 10-9–10-8 с возбужденная молекула теряет избыточную колебательную энергию и электроны переходят на основ-ной колебательный уровень возбужденного состояния (этот процесс изображен волнистой стрелкой).
При переходе с основного возбужденного уровня на какой-нибудь невозбужденный происходит излучение кванта света. Этот процесс называют флуоресценцией. Время затухания флуоресценции составляет 10-9–10-10 с.
Таким образом, флуоресценция – это излучательный переход из основного синглетного состояния на основной невозбужденный уровень.
Если происходит запрещенный по спину синглет – триплетный переход (спины параллельны), то такое излучение называют фосфоресценцией. Время жизни такого состояния велико (10-3–102 с).
Характеристики люминесценции. Люминесценция характеризуется: спектрами поглощения (зависимость А от λпогл), спектрами возбуждения (зависимость Iл от λвозб) и спектрами люминесценции (зависимость Iл от λизл). Спектры поглощения и возбуждения практически совпадают, поэтому для практических целей выбирают ту λ, при которой наблюдается наибольшее значение Iл.
Для люминесценции характерно то, что часть энергии возбуждения неизбежно теряется в виде тепла. Поэтому энергия квантов света, выделяющихся при люминесценции, меньше, чем энергия квантов возбуждающего света. Поэтому Ел < Eпогл, а λл > λ погл. Эта зависимость известна как закон Стокса: спектр люминесценции всегда смещен в
сторону более длинных волн по сравнению со спектром поглощения.
Строгое выполнение правила Стокса наблюдается лишь у атомов и простых молекул, находящихся в газовой фазе. Та часть люминесценции, где происходит нарушение правила Стокса, называется антистоксовой областью. Чтобы устранить несоответствие правила Стокса многочисленным экспериментальным данным, Ломмель предложил заменить его более широкой формулировкой: спектр излучения в целом и его максимум
всегда сдвинуты по сравнению со спектром поглощения и его максимумом в сторону длинных волн. Эта зависимость получила название закона Стокса – Ломмеля, который может быть графически представлен на риунке 4.7.27.


828
люминесценции.
Отношение излучаемой энергии люминесценции Ел к энергии поглощенного света Еп называют энергетическим выходом люминес-ценции
Вэн.
(4.7.42)
Отношение числа излученных квантов Nл к числу поглощенных квантов Nп называют квантовым выходом люминесценции Вкв.
(4.7.43)
Если учесть, что энергия квантов равна Е = N • h • ν (N – число квантов), то легко установить связь между Вэн и Вкв.
(4.7.44)
Зависимость энергетического выхода люминесценции от длины волны возбуждающего света подчиняется закону Вавилова, в соответствии с которым энергетический выход люминесценции первоначально растет
пропорционально длине волны возбуждающего света, затем в некотором интервале длин волн остается постоянным, после чего резко падает.
Согласно закону Вавилова, квантовый выход постоянен в широких пределах длины волны возбуждающего света в стоксовой области падает, если длина волны возбуждающего света лежит в антистоксовой (длинноволновой) области спектральной полосы поглощения. Падение квантового и энергетического выходов при возбуждении светом с длиной волны, лежащей в антистоксовой области, связано с уменьшением в этой области вероятности электронного перехода на возбужденный уровень.
Закон справедлив только при изменении длины волны возбуждающего света в пределах одной электронной полосы поглощения. Если при фотовозбуждении молекулы переходят в различные электронные состояния, то квантовый выход может меняться, закон не будет выполняться. Закон Вавилова строго выполняется для веществ, обладающих свойствами молекулярной люминесценции.
Тушение люминесценции. Выход люминесценции зависит от длины волны возбуждающего света, концентрации люминесцирующего вещества, посторонних примесей и температуры. Уменьшение величины выхода под влиянием этих факторов получило название тушение люминесценции.
Концентрационное тушение возникает при больших концентрациях

830
первичный светофильтр 3, кварцевую линзу 4 и попадает в анализируемый раствор 5. Люминесцентное излучение раствора собирается кварцевыми линзами (6), проходит через вторичные светофильтры 7 и попадает на фотоэлементы 8. Светофильтры 7 пропускают излучение поглощают рассеянный свет от источника возбуждения. Приемник света (фотоэлемент) измеряет люминесцентное излучение под прямым углом к направлению возбуждающего света. Фотоэлемент преобразует световую энергию в электрический ток, который усиливается и регистрируется микроамперметром. Показания микроамперметра прямо пропорциональны интенсивности люминесцентного излучения и концентрации люминесцирующего вещества в растворе.
В люминесцентном анализе исследуемое вещество чаще всего освещают УФ-лучами. Среди различных источников освещения, вызывающих люминесценцию, наибольшее распространение получили газоразрядные лампы. В качестве приемников излучения в современных приборах используются фотоумножители. Приемником люминесцентного излучения также может являться глаз человека.
Использование люминесценции в анализе:
1)Качественный анализ. Люминесцентные качественные реакции очень чувствительны, особенно когда к раствору неорганических веществ добавляют органические реагенты, вызывающие яркую люминесценцию. Например, для качественного обнаружения Zn добавляют салициловую кислоту, для обнаружения Li и Al предложен 8-оксихинолин и др.
Качественный анализ основан на способности вещества в соответствующих условиях люминесцировать или, реже, гасить люминесценцию. Возникновение или исчезновение люминесценции часто наблюдается визуально.
2)Количественный анализ. Количественный анализ основан зависимости интенсивности люминесценции от концентрации вещества, т. е. на использовании соотношения
Iл = k • С.
Для расчета содержания вещества в пробе по результатам люминесцентных измерений чаще всего используют методы градуировочного графика, сравнения и добавок.
Метод градуировочного графика. Используется наиболее часто. В этом методе измеряют интенсивность люминесценции серии стандартных образцов (растворов), охватывающих весь диапазон ожидаемых содержаний определяемого вещества в пробе, и строят график зависимости интенсивности люминесценции от концентрации определяемого вещества. В идеальном случае градуировочный график должен быть линейным и проходить через начало координат. На практике он не всегда оказывается линейным в широком диапазоне содержаний определяемого вещества и редко выходит из начала координат. В аналогичных условиях измеряют
831
интенсивность люминесценции определяемого компонента и по графику находят его концентрацию в анализируемом объекте.
Выбор оптимальных условий проведения люминесцентного анализа. Метод люминесцентного анализа позволяет количественно определять не только вещества, способные к собственному свечению, но и ряд веществ, которые не способны к люминесценции. Многие органические вещества обладают собственной люминесценцией и могут быть количественно определены путем измерения интенсивности люминесценции непосредственно анализируемых растворов.
При определении неорганических веществ, большинство из которых не обладает собственной люминесценцией, предварительно проводят аналитические реакции, в результате которых образуются способные к люминесценции комплексные соединения. В том случае, когда в результате такой реакции образуется малорастворимое в воде комплексное соединение, его экстрагируют из водной фазы органическим растворителем.
При анализе раствора, в котором присутствуют мешающие анализу примеси, проводят аналитическую реакцию с определяемым ионом и образующийся комплекс экстрагируют из водной фазы в орга-ническую. Мешающие ионы остаются в водной фазе.
Полученный экстракт определяемого вещества, существующего в составе люминесцирующего комплекса, анализируют далее обычным образом.
Достоинства метода:
1.Метод может использоваться для определения почти любых элементов при их содержании ≈ 10-5 % и меньше, многих органических веществ и др.
2.Применим для определения микропримесей.
3.Большой интерес вызывает применение люминесцентных индикаторов, которые изменяют цвет или Iл в зависимости от свойств участников реакции, рН, присутствия окислителя. Например, используя в качестве индикатора морин, можно титриметрически опреде-лять Al, Ga, Zr и другие элементы.
4.Погрешность метода составляет 5–7 %.
5.Очень низкий предел обнаружения (0,05 мкг/мл и ниже).
6.Высокая селективность.
7.Простота аппаратурного оформления.
Практическое применение:
1. В медицине – диагностика различных заболеваний (рак, малярия и
др.).
2.В фармацевтике – контроль за качеством лекарственных препаратов, анализ биологически активных веществ (витаминов, анти-биотиков и др.).
3.В сельском хозяйстве и пищевой промышленности – для определения жизнеспособности семян, качества муки, определения стадии загнивания овощей и фруктов.

832
4.В алмазодобывающей промышленности – для определения качества
алмазов.
5.В бумажной промышленности – для определения качества целлюлозы.
4.7.2.7 Рефрактометрический метод анализа
Рефрактометрический анализ основан на измерении показателя преломления (рефракции) света при прохождении его через границу раздела прозрачных сред, по которому судят о природе вещества, его чистоте или содержании в растворах.
Рефракция – это явление преломления луча света на границе раздела двух сред, различных по плотности. Количественно рефракцию оценивают по углу или показателю преломления света. Рефрактометрический метод анализа – это метод, основанный на зависимости показателя преломления света от состава системы, т.е. от концентрации компонента в растворе.
Рефрактометрия основана на измерении относительных показателей преломления веществ.
Отношение синуса угла падения α к синусу угла преломления β называют относительным показателем преломления η второго вещества по отношению к первому (рисунок 4.7.30):
(4.7.47)
Если луч света переходит из вакуума или из воздуха в другую среду, то угол падения всегда больше угла преломления. При увеличении угла падения, изменяется соотношение между долями световой энергии, проходящей в другую среду и отраженной от нее.
Рисунок 4.7.30 – Схема преломления луча
833
Показатель преломления зависит от природы, плотности и концентрации веществ, типа растворителя, температуры и других факторов. Каждая среда имеет постоянный показатель преломления и, следовательно, отношение синусов углов также является постоянной величиной. Для жидкостей и твердых тел η обычно определяют относительно воздуха, а для газов – относительно вакуума. Если в качестве сред используется не воздух, а любые другие среды, то каждая из них описывается своим показателем и предельным углом преломления.
Значения η зависят от длины волны света λ и температуры, которые указывают соответственно в подстрочном и надстрочном индексах. Например, показатель преломления при 20 °С для D-линии спектра натрия (λ = 589 нм) – ηD20 или η58920.
Обычно измерение показателя преломления проводят при температуре 20 °С. Однако при измерениях в условиях другой температуры вводят поправку на температуру по формуле
ηD20 = ηDt + 0,00023(t - 20), (4.7.48)
где, ηD20 – показатель преломления при 20 °С;
ηDt – показатель преломления при температуре образца;
t – температура, при которой проводили исследование, °С.
Для воды и водных растворов при температурах 20 ± 5 °С показатель преломления изменяется практически на одну и ту же величину составляет 1,3330, поэтому в этом интервале температур для водных растворов температурную поправку вносить не нужно.
Показатель преломления при прочих постоянных условиях связан прямой пропорциональной зависимостью с концентрацией компонента в растворе и его измерение широко используется в количественном анализе.
Обычно показатели преломления жидких и твердых тел определяют на рефрактометрах. Рефрактометры предназначены для измерения показателя преломления и средней дисперсии неагрессивных жидкостей и твердых тел, содержания белка и сухих веществ в молоке и его продуктах, для определения качества или состояния различных продуктов, сырья, плодов, ягод, меда, растительных масел.
Наиболее распространенным в аналитических лабораториях является рефрактометры Аббе, принципиальная схема которого пред-ставлена на рисунке 4.7.31.

834
Рисунок 4.7.31 – Принципиальная схема рефрактометра Аббе: 1 – источник света; 2
– призмы; 3 – исследуемое вещество; 4 – шкала; 5 – окуляр
Рефрактометр Аббе позволяет применять освещение не монохроматическим, а белым светом, отградуированным под желтую линию натрия. При этом в окуляре 5 получается окрашенная граница поля. Показатель преломления исследуемого вещества в рефрактометре обычно отсчитывают непосредственно по шкале, совмещая с ней границу светлого и темного поля.
Для рефрактометрии растворов в широких диапазонах концентраций пользуются таблицами или эмпирическими формулами, важ-нейшие из которых (для растворов сахарозы, этанола и др.) утвер-ждаются международными соглашениями и лежат в основе построе-ния шкал специализированных рефрактометров для анализа промыш-ленной и сельскохозяйственной продукции.
Определение концентрации вещества методом градуировочного графика. Готовят серию стандартных растворов определяемого вещества. Измеряют показатели преломления для каждого из приготовленных растворов. По полученным данным строят градуировочный график зависимости показателя преломления от концентрации вещества в растворе. Одну каплю исследуемого раствора наносят пипеткой на поверхность призмы и снимают отсчет по шкале показателей преломления. Содержание вещества в исследуемом растворе находят по градуировочному графику.
4.7.3 Хроматография 4.7.3.1 Общие положения и понятия хроматографии
Хроматографический метод – один из наиболее универсальных методов разделения смесей веществ. Он широко используется в различных областях науки и техники. Метод применим для разделения и анализа любых жидких и газообразных смесей веществ, даже очень близких по составу и свойствам.

835
Хроматографию можно определить, как процесс, основанный на многократном повторении актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы вдоль неподвижного сорбента.
Создатель метода – русский ученый Михаил Семенович Цвет. В 1903 г. Цвет сформулировал основы хроматографического метода. В 1906–1910 гг. он детально описал этот метод, дал его теоретическое обоснование, описал аппаратуру и технику исследований, показал его применимость.
Опыт Цвета. Цвет установил, что считавшийся однородным зеленый пигмент растений хлорофилл на самом деле состоит из нескольких веществ. При пропускании экстракта зеленого листа через колонку, заполненную мелом, и промывании петролейным эфиром он получил несколько окрашенных зон, что говорило о наличии в экстракте нескольких веществ.
Этот метод он назвал хроматографией (хроматос – цвет, греч.), хотя сам указал на возможность разделения бесцветных веществ.
Сущность метода:
1. Стеклянную трубку (колонку) с небольшим отверстием внизу, предварительно закрытым тампоном из ваты, наполняют твердым пористым веществом, не растворимым в применяемом растворителе и способным к адсорбции (рисунок 4.7.32).
Рисунок 4.7.32 – Колоночный вариант хроматографического метода
2.Через колонку пропускают смесь веществ. Вследствие различной адсорбируемости вещества смеси распределяются по высоте колонки, образуя кольца – зоны веществ. Верхняя зона содержит все компоненты, вторая сверху на один компонент меньше и т.д. Самая нижняя зона содержит
вчистом виде наименее адсорбируемый компонент смеси.
3.Если через колонку пропускать чистый растворитель, то под действием растворителя адсорбированные вещества начнут перемещаться сверху вниз с различными скоростями (тем большими, чем меньше их сорбируемость).
836
4. На определенном этапе при правильно подобранном адсорбенте и растворителе происходит полное разделение веществ. В верхней части колонки сосредотачиваются наиболее, а в нижней – наименее сорбируемые вещества.
Порядок поглощения данных веществ из данного растворителя данным адсорбентом является постоянным.
Достоинства метода:
-Универсальность.
-Высокая эффективность.
-Простота выполнения.
-Несложное оборудование.
-Отсутствие химических изменений в разделяемых веществах.
-Высокая чувствительность.
Применение метода:
-Для концентрирования веществ.
-Для определения чистоты веществ.
-Для очистки.
-Для идентификации соединений.
-Для изучения состава и строения веществ.
-Главным образом – для разделения сложных смесей органических и неорганических веществ.
Классификация хроматографических методов:
По агрегатному состоянию применяемых фаз: 1. Газовая:
– газо-жидкостная;
– газо-твердая. 2. Жидкостная:
– жидкость – жидкостная;
– жидкость – твердая;
– жидкость – гелевая. По применяемой технике
1.Колоночная (разделение веществ проводится в специальных колонках).
2.Плоскостная:
–бумажная (разделение проводится на специальной бумаге);
–тонкослойная (разделение проводится в тонком объеме сорбента). По механизмам разделения
1.Адсорбционная – разделение основано на различной адсорбируемости разделяемых веществ твердым сорбентом.
2.Распределительная – на различии в растворимости разделяемых веществ в неподвижной и подвижной фазах жидких фазах или на различии в растворимости веществ в неподвижной фаза (газовая хроматография).
3.Ионообменная – на различии в способности веществ к ионному
обмену.
837
4.Проникающая – на различии в размерах или формах молекул разделяемых веществ.
5.Осадочная – на образовании различных по растворимости осадков.
4.7.3.2 Хроматограммы и основные принципы хроматографического разделения
Любому виду хроматографии присуща следующая методика:
-Подготовка аппаратуры и реактивов.
-Получение хроматограммы.
-Анализ хроматограммы.
Хроматограмма представляет собой зависимость аналитического сигнала от времени или объема раствора, прошедшего через колонку.
Существуют три основных способа получения хроматограмм.
1.Фронтальная хроматография. Это простейший по методике вариант хроматографии. Он состоит в том, что через колонку с адсорбентом непрерывно пропускают анализируемую смесь разделяемых веществ.
Раствор, вытекающий из хроматографической колонки, называется эффлюентом.
Вследствие различной сорбции веществ сначала из колонки будет вытекать растворитель, а затем наступает насыщение сорбента наименее сорбируемым веществом, и это вещество появляется в эф-флюенте. Когда сорбент насытится вторым веществом, и это вещество появляется в эффлюенте, эффлюент содержит оба этих вещества. На третьем этапе эффлюент будет содержать три вещества и т.д. Таким образом, через некоторое время состав раствора при прохождении через колонку меняться не будет.
При фронтальной хроматографии можно отделить и получить в чистом виде лишь одно наименее сорбируемое вещество. Метод применяется, например, для очистки раствора от примесей, если они сорбируются существенно лучше, чем основной компонент, или для выделения из смеси наиболее слабо сорбирующегося вещества.
2.Элюентная (проявительная) хроматография.
Хроматографическую колонку промывают элюентом – раствором вещества или растворителем, обладающим меньшей сорбируемостью, чем любое из разделяемых веществ. Затем в колонку вводят порцию анализируемой смеси, содержащей разделяемые компоненты, растворимые в элюенте. Объем раствора должен быть мал, чтобы разделяемые вещества сосредоточились в верхней части колонки. После этого через колонку непрерывно пропускают элюент (производят элюирование). Элюирующая способность элюента различна в отношении компонентов смеси, и под действием элюента перемещение разделяемых веществ происходит с различными скоростями в соответствии с их сорбируемостью. Наименее сорбируемые вещества движутся быстрее и распределяются в нижней части колонки, наиболее сорбируемые – медленнее и располагаются в верхней части колонки.
838
На определенном этапе происходит полное разделение веществ на отдельные зоны, разделенные участками чистого сорбента. В газе или растворе, вытекающем из колонки, сначала появляется компонент, наименее сорбируемый, далее – чистый растворитель, а затем компонент, более сорбируемый.
Элюентный метод дает возможность полностью разделять сложные смеси, поэтому он более эффективен и наиболее часто применяется в практике. Недостатком метода является уменьшение концентрации выходящих растворов за счет разбавления растворителем или газомносителем.
3. Вытеснительная хроматография. В этом методе анализируемую смесь компонентов вводят в колонку и промывают раствором вещества (вытеснителем), которое сорбируется лучше, чем любой из компонентов анализируемой смеси. По мере продвижения по колонке вытеснитель сначала вытесняет более сорбируемый компонент смеси, который в свою очередь начинает вытеснять менее сорбируемый компонент, тот в свою очередь вытесняет еще менее сорбируемый и т.д. В результате анализируемая смесь перемещается впереди фронта вытеснителя, и разделяемые вещества располагаются последовательно друг
матографировании не уменьшается, в отличие от элюентного метода. Судругом.
Концентрация раствора при хрощественным недостатком вытеснительного метода является возможное наложение зоны одного вещества на зону другого, поскольку зоны компонентов в этом методе не разделены зоной растворителя.
В жидкостной хроматографии подвижной фазой всегда является жидкость, а неподвижной фазой может быть жидкой, твердой или представлять собой гель. Обычно подвижная фаза проходит через колонку с неподвижной фазой только под действием силы тяжести, и процесс разделения занимает продолжительное время. Поэтому скон-труированы приборы – жидкостные хроматографы, в которых используют колонки малого диаметра, а жидкость поступает под давлением. Этот метод называют высокоэффективная (высокоскоростная) жидкостная хроматография (ВЭЖХ), которая получила широкое применение и может конкурировать только с газовой хроматографией.
Основные принципы хроматографического разделения.
Колоночная хроматография. Рассмотрим внешнюю хроматограмму двух веществ (рисунке 4.7.33). По оси Х откладывается время хроматографирования или объем эффлюента, по оси У – аналитический сигнал.
1. Высота выходной кривой (пика) h – это перпендикуляр, опущенный из максимума пика на нулевую линию. Нулевая линия – часть хроматограммы, полученная при регистрации сигнала детектора во время выхода из колонки чистой подвижной фазы.

839
2.Ширина пика μ – отрезок, отсекаемый на нулевой линии касательными к кривой в точках перегиба, или расстояние между точками контура пика на середине высоты μ0,5.
3.Сорбционная способность неподвижной фазы по отношению
разделяемым веществам характеризуется временем удерживания tR. Время удерживания tR – это время, прошедшее от момента ввода пробы в колонку до момента выхода максимума пика вещества, т.е. это время пребывания вещества в подвижной и неподвижной фазе. Это очень важная величина, так как если условия разделения (скорость потока подвижной фазы, давление, температура, состав подвижной и неподвижной фаз) постоянны, то время удерживания строго воспроизводимо и является характеристикой вещества, поэтому может быть использовано для идентификации веществ.
Рисунок 4.7.33 – Дифференциальная хроматограмма:1 – нулевая линия; 2 – пик несорбирующегося компонента; 3, 4 – пики определяемых компонентов; tR –время удерживания; h – высота пика; – ширина пика
4. Удерживающий объем VR является такой же важной характеристикой:
VR = F • tR,
(4.7.49)
где F – объемная скорость потока.
Символами tR0 и VR0 обозначают время и объем удерживания несорбирующегося компонента.
5. Разделение двух соседних пиков характеризуется разрешением. Разрешение пиков зависит от их остроты (ширина полос) и от расстояния между максимумами (разделение полос). Острота пиков зависит от эффективности колонки, а расстояние между максимумами определяется ее селективностью.
Под эффективностью колонки понимают получение узких пиков, т.е. ограничение размывания (расширения) полос. В эффективной колонке размывание полос небольшое и пики получаются узкими.
Расстояние между максимумами пиков определяется селективностью

840
колонки, т. е. селективностью сорбента и различиями в термодинамических свойствах хроматографируемых веществ по отношению к хроматографической системе. Селективность колонки зависит от констант и коэффициентов распределения компонентов смеси коэффициентов емкости колонки. При малых значениях коэффициентов компоненты слабо удерживаются колонкой, и наблюдается плохое разделение. При больших значениях коэффициентов – разделение увеличивается, но растет и время разделения.
4.7.3.3 Хроматографический метод анализа
Колоночная хроматография Качественный анализ проводят:
-Хроматографическими методами по параметрам удерживания (времени удерживания и удерживаемому объему). Для этого сравнивают эти характеристики в тех же условиях с такими же характеристиками стандартных или известных веществ.
-Нехроматографическими методами. Идентифицируют компоненты или вещества, используя ИК-, ЯМР-, масс-спектроскопию и другие методы анализа.
Количественный анализ проводят, измеряя высоту или площадь пика, так как эти параметры пропорциональны концентрации вещества или его количеству в хроматографической зоне. Высота пика используется только тогда, когда время удерживания малое (пик острый) и форма пика не искажена (высота пика изменяется линейно). Поэтому площадь пика используется чаще.
Для расчета хроматограмм используют несколько методов:
-нормировки (метод внутренней нормализации);
-внешнего стандарта (градуировочного графика);
-внутреннего стандарта.
Метод нормировки (метод внутренней нормализации):
1.Регистрируют пики для каждого компонента (например, x, y, z).
2.Доля площади каждого пика соответствует доле компонента в пробе, например, для компонента х:
(4.7.50)
где S – площади пиков соответствующих компонентов.
Если известен поправочный коэффициент k, определяемый чувствительностью детектора хроматографа к компоненту, то расчет проводят по формуле
841
(4.7.51)
Метод внешнего стандарта (метод градуировочного графика):
1.Готовят стандартные растворы для определяемых компонентов.
2.Получают хроматограммы.
3.Рассчитывают площадь пика (измеряют высоту пика) для каждого раствора.
4.Строят график зависимости площади пика (высоты) от концентрации компонента.
5.Получают хроматограмму определяемого компонента, рассчитывают площадь пика (высоту) и по графику определяют его содержание в пробе.
Метод внутреннего стандарта:
1.В анализируемую смесь вводят определенное количество стандартного вещества (внутреннего стандарта).
2.Расчеты проводят по формуле
(4.7.52)
где Sст – площадь пика вещества, введенного в качестве внутреннего стандарта;
kст – его поправочный коэффициент;
R – отношение массы внутреннего стандарта к массе анализируемой пробы.
Графический вариант. Составляют смеси точного состава внутреннего стандарта с каждым из компонентов. При этом используют различные соотношения внутреннего стандарта и компонентов. Строят график зависимости площади пиков S от процентного содержания компонентов. По графику определяют содержание компонента.
Осадочная хроматография. Качественный анализ. Если зоны хроматограммы окрашены, то по их числу, окраске и расположению судят о качественном составе анализируемой смеси. Если хроматограмма бесцветна, то используют раствор проявителя, образующего окрашенные соединения с разделяемыми ионами.
Количественный анализ. Используют зависимость высоты зоны хроматограммы от концентрации вещества.
Бумажная распределительная хроматография. Для разделения компонентов используют специальную бумагу, на которую наносят анализируемый раствор. Затем бумагу помещают герметичную камеру и один ее конец погружают в растворитель, который является подвижной фазой. Под действием капиллярных сил растворитель движется по бумаге, растворяя и увлекая за собой компоненты образца. До начала движения
842
образец должен полностью раствориться, поэтому скорость растворения компонентов в подвижной фазе является одним из факторов, определяющих эффективность разделения. После того, как растворитель пройдет определенное расстояние, лист вынимают и сушат. Затем образовавшиеся пятна, которые могут быть как видимые, так и невидимые, обнаруживают и отмечают. Если хроматографические зоны бесцветны, то бумагу подсушивают и опрыскивают проявителем.
Качественный анализ проводят по характерной окраске зон. Количественный анализ – сравнением интенсивности окраски и величины зоны со стандартными растворами.