

37. Миеломная болезнь. Определение, распространенность, этиопатогенез, классификация, клиническая картина. Критерии диагностики. Лечение. Прогноз.
Миеломная болезнь - злокачественное В-лимфопролиферативное заболевание, субстратом которого являются опухолевые плазматические клетки, проявляющееся парапротеинемией, костномозговой недостаточностью, костными деструкциями, гиперкальциемией, почечной недостаточностью и рецидивирующей вторичной инфекцией
Болезнь встречается чаще в возрасте от 40 до 70 лет. Частота ее составляет 1:100тыс.
Этиопатогенез
Длительная, хроническая антигенная стимуляция после вирусных инфекций или других хронических заболеваний, длительного воздействия токсических веществ и радиации
В результате длительной серии генетических событий формируется патологическский клон В-клеток, способных к дифференцировке до плазматических клеток, но продуцирующих нефункциональный иммуноглобулин
Классификация
Стадии: 1 стадия:
1. Hb >100 г/л. 2. Кальций сыворотки крови <12 мг/дл. 3. Рентгенологическое поражение костей — норма или имеет место солитарное поражение. 4. Продукция М-градиента на низком уровне: – уровень IgG<50 г/л; – уровень IgА <30 г/л. 5. Экскреция легких цепей
2 стадия: Критерии не соответствуют ни I, ни III стадиям ≥ 30 г/л ≥ 10 г/л ≥ 1 г
3 стадия: Один или более из нижеперечисленных критериев:
1
 .
Hb <85 г/л.
2. Кальций сыворотки крови
>12 мг/дл.
3. Рентгенологическое
поражение костей — выраженные костные
деструкции.
4. Продукция М-градиента
на высоком уровне:
– уровень IgG >70
г/л;
– уровень IgА >50 г/л.
5. Экскреция
легких цепей >12 г/24 ч
Клиническая
картина
.
Hb <85 г/л.
2. Кальций сыворотки крови
>12 мг/дл.
3. Рентгенологическое
поражение костей — выраженные костные
деструкции.
4. Продукция М-градиента
на высоком уровне:
– уровень IgG >70
г/л;
– уровень IgА >50 г/л.
5. Экскреция
легких цепей >12 г/24 ч
Клиническая
картина
1. Болевой синдром, связанный с костными деструкциями. Наиболее типичны люмбалгии.
2. Признаки хронической почечной недостаточности вследствие миеломной нефропатии.
3. Анемический синдром.
4. Частые инфекционные заболевания из-за развития иммунодефицита (синдром недостаточности антител).
5. Гиперкальциемия, вызывающая слабость, жажду и тошноту, острую почечную недостаточность.
6. Гипервязкость крови с развитием кровоточивости и различных ишемических неврологических нарушений (парапротеинемическая кома).
7. Амилоидоз.
Критерии диагностики
Диагноз может быть поставлен при наличии двух критериев из трех: 1. Содержание плазматических клеток в костном мозге ≥10%. 2. Концентрация моноклонального белка в сыворотке крови≥30 г/л (IgG), ≥10 г/л (IgА) или экскреция легких цепей иммуноглобулинов с мочой (каппа или лямбда) ≥1 г/24 ч. 3. Остеолитическое поражение костей.
План обследования:
Клинический анализ, б/х анализ крови, общий анализ мочи. При протеинурии — определение суточной потери белка с мочой, электрофорез белков мочи. Иммуноферментный анализ сыворотки крови и мочи на моноклональный иммуноглобулин и легкие цепи. Стернальная пункция для морфологического исследования костного мозга (миелограмма).
Трепанобиопсия для гистологического исследования костного мозга. Рентгенограммы костей скелета (плоские кости, позвоночник, при необходимости — трубчатые кости) или изотопная сцинтиграфия скелета. Для уточнения класса и типа моноклонального иммуноглобулина, определения количества нормальных иммуноглобулинов — иммуноэлектрофорез (качественное определение) и радиальная иммунодиффузия (количественное определение) с моноспецифическими антисыворотками против тяжелых и легких цепей (метод иммунофиксации)
Лечение
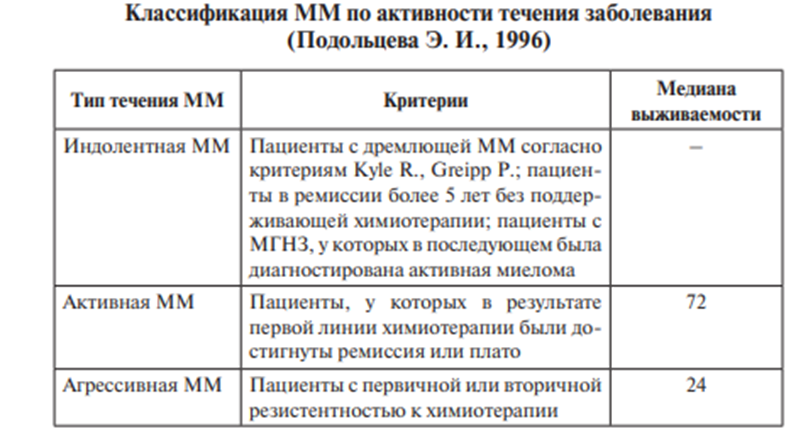
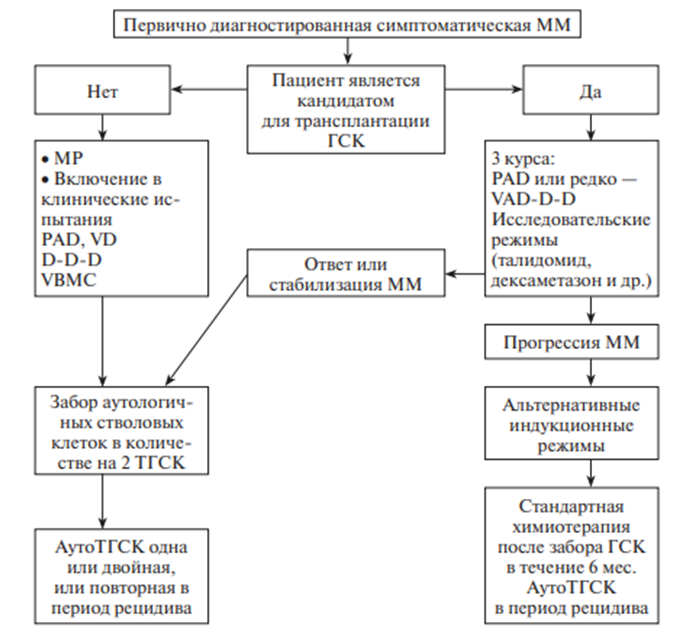
За основу берется тип течения заболевания - больным с индолентным и дремлющим течением ММ химиотерапия не проводится, показано динамическое наблюдение. В случае прогрессии заболевания назначается специфическая терапия
В терапии используется препарат ВЕЛКЕЙД (бортезамиб) – ингибитор протеасом плазмоцитов, что стимулирует апоптоз PAD: Велкейд + дексаметазон + адриабластин
Агрессивное
течение ММ является показанием для
интенсификации лечения по протоколу
РАD и далее — проведения высокодозной
химиотерапии с поддержкой аутологичными
стволовыми клетками периферической
крови или костного мозга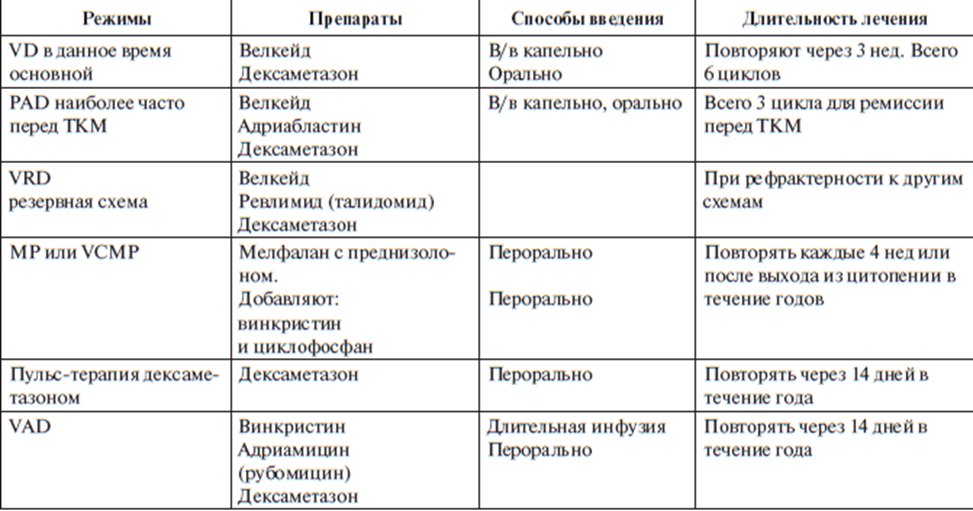
38. Хронические миелопролиферативные заболевания: хронический миелолейкоз, идиопатический миелофиброз, полицитемия. Распространенность, клиническая картина, диагностика. Дифференциальная диагностика.
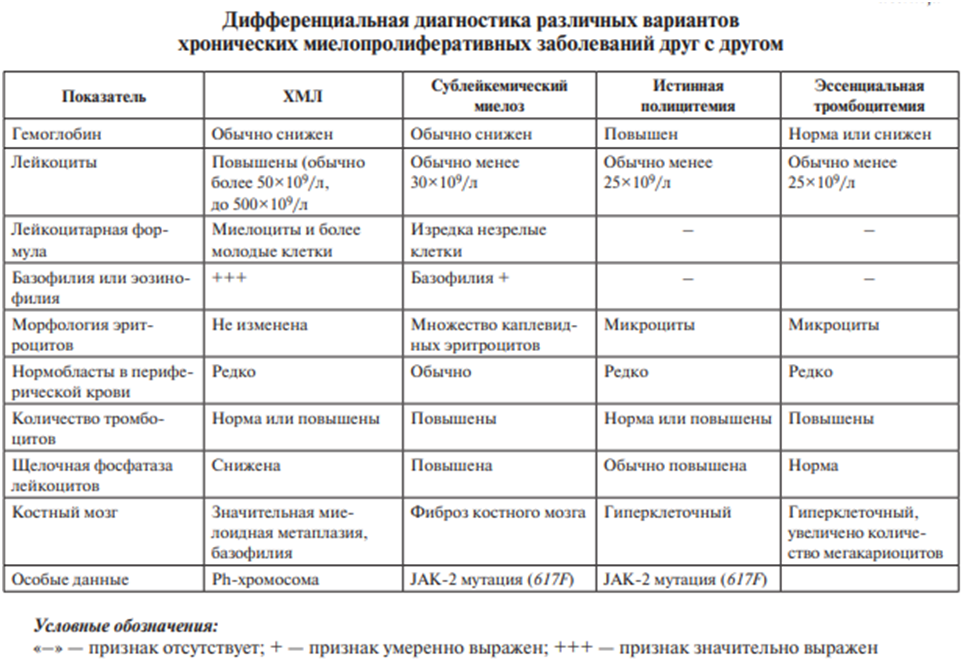
Хронический миелолейкоз (ХМЛ) — опухолевое заболевание системы крови, возникающее из ранних предшественников миелопоэза, морфологическим субстратом которого являются преимущественно созревающие и зрелые гранулоциты, в основном нейтрофилы.
Истинная полицитемия — хроническое миелопролиферативное опухолевое заболевание, при котором наблюдается увеличение массы циркулирующих эритроцитов за счет повышения их продукции в костном мозге.
Сублейкемический миелоз — хроническое миелопролиферативное заболевание, характеризующееся фиброзом костного мозга, развитием экстрамедуллярного кроветворения в печени и селезенке, с картиной периферической крови, подобной ХМЛ.
Клиническая картина.
Течение хронического миелолейкоза носит прогрессирующий характер, в соответствии с этим выделяют три фазы заболевания:
1. Хроническая фаза, которая в большинстве случаев протекает бессимптомно. Иногда больные жалуются на необоснованную утомляемость, снижение трудоспособности. В клиническом анализе крови выявляется нейтрофильный лейкоцитоз, часто увеличено количество базофилов и эозинофилов (базофильно-эозинофильная ассоциация). На ранних этапах заболевания анемии, тромбоцитопении обычно не наблюдается. При исследовании миелограммы обращает на себя внимание увеличение количества миелокариоцитов, в основном за счет незрелых форм гранулоцитов: метамиелоцитов и миелоцитов, промиелоцитов, единичных бластов. При просмотре мазков выявляется увеличение числа мегакариоцитов и свободно лежащих тромбоцитов. При трепанобиопсии костного мозга выявляется рассасывание костной ткани, заметное уменьшение числа жировых клеток, вплоть до полного их исчезновения вследствие нарастания элементов гранулопоэза, с преобладанием среди них незрелых форм. При биохимическом исследовании обнаруживается снижение активности щелочной фосфатазы нейтрофилов. Единственным доказательством ХМЛ является проведение цитогенетического исследования: обнаружение специфической хромосомной аномалии в гемопоэтических клетках — филадельфийской хромосомы (Ph-хромосома, t (9:22), или химерного гена bcr/abl).
2. Фаза акселерации характеризуется развитием симптомов опухолевой интоксикации (общая слабость, потливость, потеря веса, персистирующая лихорадка, не связанная с инфекцией), тяжестью, болями в левом подреберье, обусловленными нарастающей спленомегалией, оссалгиями. Критериями фазы акселерации считается: увеличение содержания бластов более 10% или суммарное содержание бластов и промиелоцитов более 30% в периферической крови и костном мозге; увеличение количества базофилов и эозинофилов более 10% в костном мозге; возможно появление тромбоцитопении менее 100×109/л, цитогенетическая клональная эволюция заболевания (появление дополнительной Ph-хромосомы, трисомия 8-й пары, изохромосомия длинного плеча 17-й хромосомы и др.). В фазу акселерации прогрессивно увеличивается клеточность костного мозга, время удвоения количества лейкоцитов сокращается до 20 дней и менее.
3. При бластном кризе общее состояние пациента резко ухудшается. Значительно выражены признаки опухолевой интоксикации. Появляются экстрамедуллярные очаги лейкозного роста: сильные боли в костях вследствие развития поднадкостничных лейкозных инфильтратов, боли в животе из-за увеличения печени и селезенки, обусловленного их лейкозной инфильтрацией, развитием инфарктов в селезенке и периспленитов, увеличение лимфатических узлов, кожные лейкемиды. Бластный криз характеризуется: увеличением количества бластов в костном мозге или периферической крови более 20% и возникновением очагов экстрамедуллярного опухолевого кроветворения. Различают миелобластный, лимфобластный вариант криза, но бластный криз может характеризоваться пролиферацией эритробластов, монобластов или морфологически недифференцируемых бластов.
Сублейкемический миелоз (идиопатический миелофиброз) наиболее часто встречается в возрастной группе 50–70 лет и долго остается бессимптомным. Первым симптомом, как правило, является спленомегалия, которая может постепенно достигать гигантских размеров, нередко сопровождается болевым синдромом в области левого подреберья вследствие инфаркта селезенки. Картина периферической крови и аспирата костного мозга в первые фазы заболевания напоминает таковую при ХМЛ.
Критерием установления диагноза является: 1. Миелофиброз, выявляемый при гистологическом исследовании трепанобиоптата костного мозга. 2. Отсутствие филадельфийской хромосомы в гемопоэтических клетках при цитогенетическом исследовании и гена bcr/abl в крови, наличие мутации гена JAK-2 (617F) в крови, высокий уровень щелочной фосфатазы в нейтрофилах при гистохимическом исследовании.
Истинная полицитемия — относительно редкое заболевание с длительным, хроническим течением. Средний возраст больных около 60 лет. Основные клинические симптомы — слабость, быстрая утомляемость, кожный зуд, плетора, эритромелалгия, картина тромбозов различных локализаций, кровоточивость, обусловленная нарушением функции тромбоцитов. В клиническом анализе крови выявляются эритроцитоз, часто би- или панцитоз (эритроцитоз, лейкоцитоз, тромбоцитоз), в трепанобиоптате костного мозга — трехростковая гиперплазия.
Основными диагностическими критериями являются:
1. Увеличение массы циркулирующих эритроцитов, не зависящее от уровня эритропоэтина.
2. Трепанобиопсия подвздошной кости с уменьшением или исчезновением жировой ткани за счет пролиферации деятельного костного мозга.
3. Спленомегалия.
4. Наличие в крови и костном мозге мутации гена JAK-2 (617F).
Также учитываются такие признаки, как тромбоцитоз более 400×109/л, лейкоцитоз более 10×109/л, повышение уровня щелочной фосфатазы нейтрофилов.
В ряде случаев возникает необходимость дифференциальной диагностики с острым лейкозом: клинические признаки не позволяют надежно различить бластный криз ХМЛ и острый лейкоз. Следует помнить, что для острого лейкоза характерен разрыв, провал в миелограмме между бластными клетками и зрелыми элементами: резкое повышение бластов не сопровождается ростом числа промежуточных форм — промиелоцитов и миелоцитов, как это бывает при хроническом миелолейкозе. От ХМЛ приходится дифференцировать лейкемоидные реакции, развивающиеся при различных заболеваниях.
При постановке диагноза эссенциальной тромбоцитемии следует исключить все возможные причины тромбоцитоза:
– кровотечения;
– солидные опухоли;
– другие миелопролиферативные заболевания (ХМЛ, сублейкемический миелоз, истинная полицетемия);
– инфекции.