

Биологически важные реакции нуклеофильного замещения с участием эфиров фосфорной кислоты:
1. Перенос фосфатных групп.
2. Синтез нуклеиновых кислот.
3. Сигнальные пути.
4. Метаболизм.
Внутри- и межмолекулярная дегидратация спиртов (образование алкенов и простых эфиров). Окисление первичных и вторичных спиртов.
При нагревании спиртов в присутствии концентрированной серной кислоты, безводной фосфорной кислоты или при пропускании паров спирта над катализатором алюминия оксидом Al2O3 спирты отщепляют воду, то есть подвергаются дегидратации. в зависимости от природы спирта и условий проведения реакции дегидратация может протекать межмолекулярно и внутримолекулярно. При межмолекулярной дегидратации спиртов образуются простые эфиры.
В результате внутримолекулярной дегидратации образуются алкены.
Внутримолекулярная дегидратация в ряду вторичных и третичных спиртов протекает согласно правилу Зайцева: протон отщепляется от соседнего, менее гидрогенизированного атома углерода.
Межмолекулярная и внутримолекулярная дегидратации спиртов представляют собой два конкурирующих процесса, из которых каждый в определенных условиях может стать доминирующим. отщепление воды от двух молекул спирта с образованием простых эфиров — межмолекулярная дегидратация — становится преобладающим процессом при нагревании спиртов в присутствии каталитических количеств минеральной кислоты (спирт в избытке) при температуре 140—160 °С. Внутримолекулярная дегидратация, то есть превращение спирта в алкен, становится доминирующей при нагревании спиртов с избытком минеральной кислоты при температуре выше 170 °С. особенно легко она протекает в ряду третичных спиртов. Межмолекулярная дегидратация спиртов протекает по механизму SN2 или SN1. При этом вначале молекула спирта под действием минеральной кислоты протонируется с образованием оксониевого катиона, а затем происходит замещение группы —OH. Механизм SN2 включает образование переходного состояния, которое формируется в процессе нуклеофильной атаки электрофильного атома углерода оксониевого катиона второй молекулой спирта.
Первичные и вторичные спирты по-разному относятся к действию окислителей. Первичные спирты при окислении первоначально образуют альдегиды, которые могут окисляться далее, превращаясь при этом в карбоновые кислоты. Вторичные спирты при окислении образуют кетоны. В качестве окислителей для окисления спиртов используют хрома (VI) оксид, калия дихромат в серной кислоте (хромовая смесь), калия перманганат в серной кислоте и др. В промышленности для окисления первичных спиртов в альдегиды используют метод каталитического дегидрирования. сущность метода состоит в пропускании паров спирта над катализатором (мелкораздробленная медь) при 280—300 °с. Происходит отщепление молекулы водорода от молекулы спирта и образуется альдегид.
Преимуществом каталитического дегидрирования является то, что предотвращается более глубокое окисление альдегида до кислоты, в условиях данной реакции из вторичных спиртов синтезируют и многие кетоны.
Многоатомные спирты. Этиленгликоль, глицерин, пентаэритрит, инозит. Химические свойства 1,2-диолов: кислотность, образование хелатных комплексов, окислительное расщепление 1,2-диолов (йодной кислотой), образование циклических простых и сложных эфиров азотной кислоты.
Двухатомные спирты (содержат две гидроксильные группы) называют диолами или гликолями. По систематической номенклатуре IUPAC названия гликолей образуют, исходя из названия соответствующего углеводорода, добавляя суффикс -диол и цифровые локанты, указывающие положение гидроксильных групп в углеродной цепи. Трехатомные спирты (содержат три гидроксильные группы) называют триолами,или глицеринами. По заместительной номенклатуре названия трехатомных спиртов образуют путем добавления к названию соответствующего углеводорода суффикса –триол. Многоатомные спирты содержат более трех гидроксильных групп и их называют полиолами. Так, четырехатомные спирты имеют общее название «эритриты», пятиатомные — «пентиты», шестиатомные — «гекситы» и т. д. Низшие члены гомологического ряда диолов представляют собой вязкие жидкости, высшие — кристаллические вещества. Жидкие гликоли имеют большую плотность и более высокие температуры плавления и кипения, чем одноатомные спирты; хорошо растворяются в воде. трехатомные спирты — вязкие жидкости или трудно кристаллизующиеся твердые вещества. Вязкость, растворимость в воде, температуры плавления и кипения гидроксильных производных алифатических углеводородов увеличиваются в ряду: одноатомные спирты < гликоли < глицерины. Такая зависимость является следствием усиления ассоциации молекул за счет образования межмолекулярных водородных связей. Отличительной особенностью гидроксильных производных углеводородов с несколькими он-группами является их сладковатый вкус, как правило, усиливающийся с увеличением числа гидроксильных групп в молекуле.
Этиленгликоль (1,2-этандиол) но–сн2–сн2–он. бесцветная вязкая жидкость, т. кип. 197,6 °с, т. пл. –11,5 °с. Гигроскопичен, смешивается с водой и этанолом, сильно понижает температуру замерзания воды и используется для приготовления антифриза. очень токсичен. широко используется для получения синтетических волокон.
Глицерин (1,2,3-пропантриол). бесцветная сиропообразная жидкость без запаха, со сладким вкусом, т. пл. 18 °с, т. кип. 290 °с (с разложением). Гигроскопичен, смешивается с водой и этанолом в любых соотношениях. Применяется в качестве основы для мазей и паст, добавки к мылам. в больших количествах глицерин используется для получения нитроглицерина.
Пентаэритрит (2,2-бис(гидроксиметил)пропан-1,3-диол) C(CH2OH)4 — четырёхатомный спирт с углеродным скелетом неопентана. Белый кристаллический порошок со сладким вкусом. Пентаэритрит проявляет свойства спиртов: алкилируется, ацилируется, реагирует с азотной кислотой с образованием моно-, ди-, три- и тетрапроизводных; образует алкоголяты и комплексы с металлами. Отличительной чертой пентаэритрита является способность образовывать циклические производные. Так, при реакции с тионилхлоридом, в зависимости от условий, образует моно-, ди-, трихлорпроизводное или пентаэритритдисульфит. Пентаэритрит применяется в производстве алкидных смол, пентафталевых лаков и эмалей, синтетических смазочных масел, пентапласта, пластификаторов и антиоксидантов для полимеров, термостабилизаторов, для синтеза ПАВ, взрывчатого вещества тетранитропентаэритрита (ТЭНа).
Инозитол (циклогексан-1,2,3,4,5,6-гексол) — шестиатомный спирт циклогексана. Более точное название — мио-инозитол. Представляет собой карбоциклический сахар, который содержится в большом количестве в мозге и других тканях млекопитающих, опосредует передачу клеточного сигнала в ответ на различные гормоны, нейротрансмиттеры и факторы роста и участвует в осморегуляции. Инозитол существует в девяти стереоизомерах, из которых наиболее часто встречающимся в живых организмах является цис-1,2,3,5-транс-4,6-циклогексангексаол. Несмотря на сходную с сахаридами брутто-формулу Cx(H2O)y, инозитол по химической природе не является углеводом, он практически безвкусный, слегка сладкий. Инозитол называли «витамином В8», однако было показано, что около 3/4 суточной потребности инозитола вырабатывается самим организмом, поэтому инозитол относят к витаминоподобным веществам. Не существует данных о том, что недостаток инозитола в пище может вызывать болезненные проявления. Двухатомные спирты вступают в те же реакции, что и одноатомные, с той лишь разницей, что они могут протекать с участием одной или двух гидроксильных групп. еще большее разнообразие продуктов возможно для реакций с участием трех- и полиатомных спиртов. При взаимодействии гликолей со спиртами, минеральными или органическими кислотами образуется два ряда производных: а) неполные и полные простые эфиры; б) неполные и полные сложные эфиры. При взаимодействии глицерина с концентрированной азотной кислотой в присутствии концентрированной серной кислоты получают полный азотнокислый эфир глицерина — глицерина тринитрат (нитроглицерин).
При окислении гликолей образуется смесь продуктов окисления:
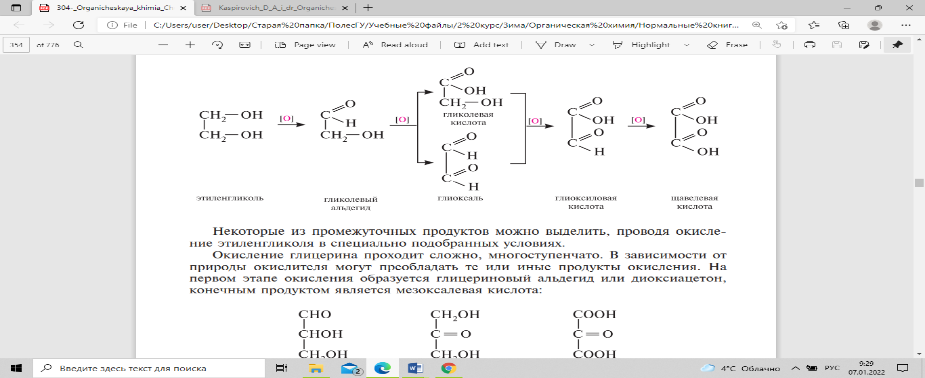
Некоторые из промежуточных продуктов можно выделить, проводя окисление этиленгликоля в специально подобранных условиях.
Окисление глицерина проходит сложно, многоступенчато. В зависимости от природы окислителя могут преобладать те или иные продукты окисления. Специфической реакцией окисления 1,2-диолов является их взаимодействие с йодной кислотой НIO4 (гликольное расщепление). В процессе реакции происходит расщепление углерод-углеродной связи в диольном фрагменте, при котором в зависимости от строения гликоля образуются соответствующие альдегиды и кетоны:

В эту же реакцию вступают и глицерины. Многоатомные спирты, содержащие гидроксильные группы у соседних атомов углерода, при взаимодействии с гидроксидами тяжелых металлов, например гидроксидом меди (II) в щелочной среде, образуют внутрикомплексные, так называемые хелатные, соединения. Такие соединения обычно хорошо растворимы в воде и интенсивно окрашены, поэтому реакция используется как качественная. При взаимодействии этиленгликоля или глицерина с гидроксидом меди (II) возникает интенсивно синее окрашивание в результате образования гликолята меди (II) или глицерата меди (II).

Эта качественная реакция характерна для многоатомных спиртов с открытой цепью и некоторых циклических спиртов, в которых гидроксильные группы достаточно сближены.
Фенолы. Номенклатура и изомерия. Простейшие представители: фенол, крезолы, пирокатехин, резорцин, гидрохинон, флороглюцин, пирогаллол. Электронное строение фенола. Кислотность фенолов.
Фенолами (аренолами) называют производные ароматических углеводородов, содержащие одну или несколько гидроксильных групп, непосредственно связанных с атомами углерода ароматического цикла. Название класса произошло от простейшего представителя — фенола с6н5он. Фенолы — устойчивые соединения и существуют исключительно в енольной форме.
Большая стабильность енольной формы обусловливается ароматическим строением углеводородного радикала и высокой степенью сопряжения он-группы с бензольным ядром. Фенолы существенно отличаются по своим физическим и химическим свойствам и от спиртов. Главной причиной этих отличий является различный характер электронных взаимодействий гидроксильной группы с углеводородным радикалом. В спиртах гидроксильная группа связана с атомом углерода в sp3-гибридизации. В фенолах кислородный атом гидроксильной группы связан с атомом углерода ароматической системы и поэтому наряду с отрицательным индуктивным эффектом имеет место и положительный мезомерный эффект. В результате мезомерного эффекта происходит смещение неподеленной пары электронов атома кислорода к бензольному кольцу и на кислороде возникает частичный положительный заряд. такой же по величине отрицательный заряд переходит на ароматическое ядро. Так как для гидроксильной группы мезомерный эффект по силе превосходит индуктивный, то суммарный частичный заряд на атоме кислорода фенольного гидроксила положителен, тогда как кислородный атом спиртового гидроксила имеет частичный отрицательный заряд.
По заместительной номенклатуре IUPAC названия фенолов образуют от названий соответствующих аренов с добавлением префикса гидрокси-. Для многих фенолов используют тривиальные названия. В качестве основы названий гомологов фенола чаще всего используют слово «фенол».
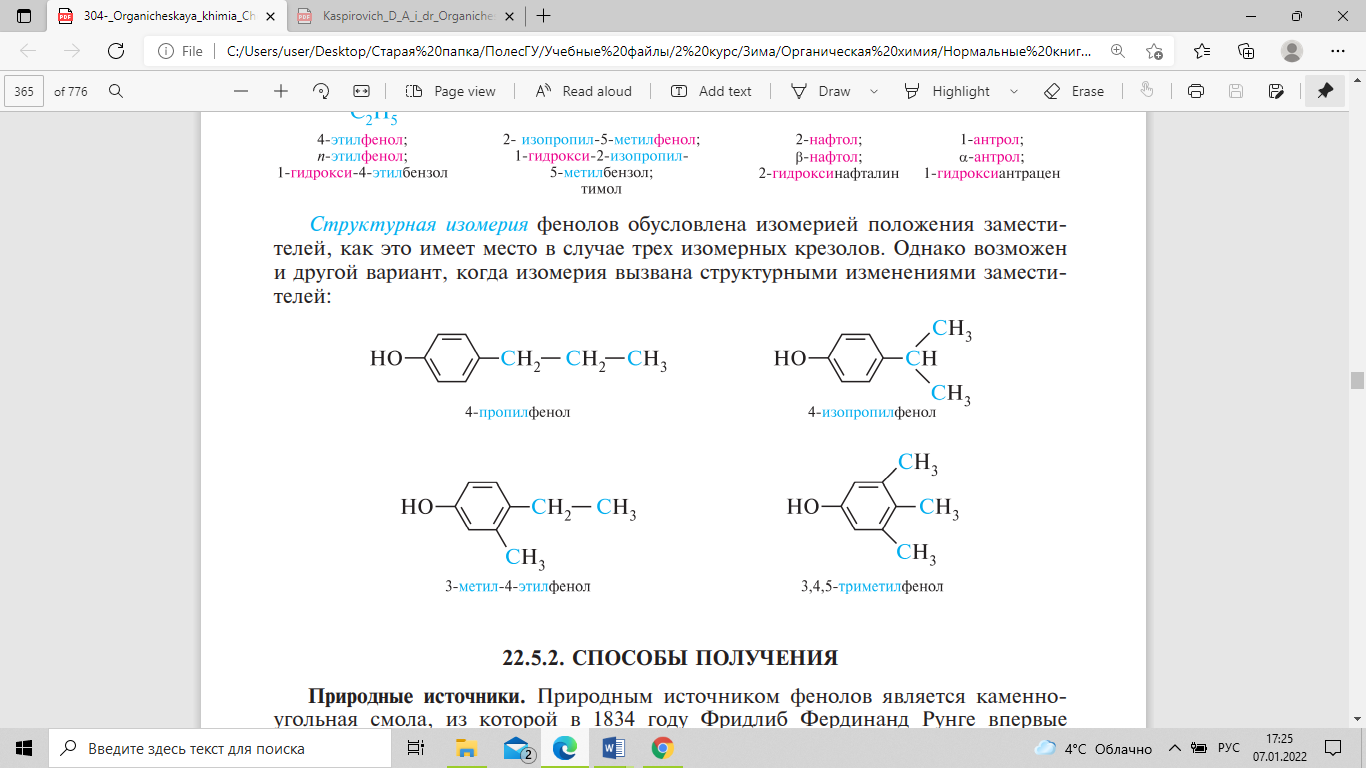
Структурная изомерия фенолов обусловлена изомерией положения заместителей, как это имеет место в случае трех изомерных крезолов. Однако возможен и другой вариант, когда изомерия вызвана структурными изменениями заместителей:
Фенол с6н5он. бесцветные, розовеющие на воздухе вследствие окисления кристаллы (т. пл. 43 °с, т. кип. 182 °с). Растворяется в воде (при 15 °с — около 8 %). Обладает антисептическими свойствами. В виде 5 %-ного водного раствора (карболовая кислота) используется в качестве дезинфицирующего средства. Нашел широкое применение в производстве пластмасс, красителей, взрывчатых веществ, лекарственных средств. Фенол токсичен, может вызвать ожоги кожи. Фенолы — более сильные он-кислоты, чем спирты. Это вызвано тем, что неподеленная пара электронов атома кислорода в молекуле фенола смещена к ядру (+М-эффект он-группы), что приводит к образованию частичного положительного заряда на атоме кислорода и увеличению поляризации связи о—н по сравнению со спиртами. кроме того, образующийся фенолят-ион (феноксид-ион) имеет повышенную стабильность за счет делокализации отрицательного заряда по ароматическому радикалу. Доказательством более сильных кислотных свойств фенолов по сравнению со спиртами может служить их реакция с водными растворами щелочей с образованием солей — фенолятов (феноксидов).
Крезолы (о-, м-, п-метилфенолы). Применяются для получения пластмасс, красителей и др. смесь изомерных крезолов с мылами используется как дезинфицирующее средство в ветеринарной практике (лизол, креолин).
Пирокатехин (1,2-дигидроксибензол) — органическое соединение, двухатомный фенол, имеющий химическую формулу С6Н4(ОН)2. Один из трёх возможных изомеров дигидроксибензола, два других — гидрохинон и резорцин. Выглядит как бесцветные кристаллы с запахом фенола. Сильный восстановитель. Применяют в фотографии как проявитель, в производстве красителей, лекарственных веществ (например, адреналина). Этерифицированием из пирокатехина получают гваякол, — исходное вещество для синтеза ванилина.
Гидрохинон (пара-дигидроксибензол, бензол-1,4-диол, хинол) — ароматическое органическое соединение, представитель двуатомных фенолов с химической формулой C6H4(OH)2, изомер пирокатехина и резорцина. Название «гидрохинон» дано соединению Ф. Вёлером из-за схожести свойств с хиноном, из которого Вёлер его синтезировал. Сильный восстановитель, используется как проявитель в фотографии, антиоксидант в химической промышленности, реагент для определения ниобия, вольфрама, золота и цезия в аналитической химии. Обладает слабым дезинфекционным действием, аналогичным тому, который оказывает фенол.
Флороглюцин (1,3,5-тригидроксибензол) — трёхатомный фенол. Представляет собой бесцветные кристаллы, сладкие на вкус. Растворим в этиловом спирте, эфире, ацетоне, трихлорметане, пиридине. В воде растворим слабо (1,13 г/100 мл при 35 °C). Образует дигидрат с температурой плавления 116—117 °C. Безводный флороглюцин плавится при 223 °C, при дальнейшем повышении температуры возгоняется с разложением. Проявляет кето-енольную таутомерию. Водным раствором карбоната (или гидрокарбоната) натрия при комнатной температуре карбоксилируется до 2,4,6-тригидроксибензойной (флороглюцинкарбоновой) кислоты. Применяют для качественного и количественного определения пентоз и пентозанов. Используется в фотоэмульсиях, как вулканизирующий реагент для каучуков, в синтезе лекарственных препаратов.
Пирогаллол (пирогалловая кислота, 1,2,3-тригидроксибензол) — органическое соединение, трехатомный фенол с химической формулой C6H6O3, бесцветные кристаллы, темнеющие на воздухе. Применяется в органическом синтезе как восстановитель, также используется в промышленности как полупродукт в производстве красителей, в фотографии как проявляющее вещество.
На кислотность фенола значительное влияние оказывают заместители в ароматическом ядре. Так, введение в n-положение бензольного ядра молекулы фенола электроноакцепторных заместителей (—NO2, —CN, —Hal и др.) усиливает кислотные свойства фенола. введение же в n-положение электронодонорных заместителей (—NH2, —OCH3 и др.) приводит к понижению кислотности, поскольку при этом уменьшается смещение электронов связи O—H к атому кислорода, что затрудняет отрыв протона.
Образование простых и сложных эфиров фенолов. Реакции электрофильного Замещения в ряду фенолов (галогенирование, сульфирование, нитрование, алкилирование).
При взаимодействии фенолятов с галогеналканами образуются простые эфиры. Такие реакции называют O-алкилированием, так как при этом алкилируется атом кислорода. Метиловые эфиры фенолов обычно получают взаимодействием фенолятов с диметилсульфатом (о-алкилирование).
Аналогично протекает реакция образования сложных эфиров при взаимодействии фенолят-иона с ацилирующими реагентами — галогенангидридами и ангидридами карбоновых кислот (о-ацилирование): Ацилирование фенолов карбоновыми кислотами в присутствии серной кислоты, в отличие от ацилирования спиртов, практически не используется, так как эта реакция из-за уменьшения электронной плотности на атоме кислорода в молекуле фенола идет значительно медленнее. Гидроксильная группа, проявляя электронодонорные свойства, очень сильно активирует бензольное кольцо по отношению к электрофильным реагентам. Фенолят-ионы в реакции SE еще более активны, чем соответствующие фенолы. В связи с высокой активностью фенолов необходимо принимать специальные меры для того, чтобы предотвратить реакции окисления и полизамещения. Обычно для введения атома галогена в бензольное кольцо требуются катализаторы — кислоты Льюиса. Реакция галогенирования фенолов, учитывая их высокую реакционную способность, протекает очень легко в отсутствие катализатора. они обесцвечивают бромную воду, причем происходит замещение всех атомов водорода в о- и п-положениях. Дальнейшее бромирование приводит к образованию так называемого «тетрабромфенола». При этом происходит нарушение ароматичности бензольного кольца. Для введения в молекулу фенола одного или двух атомов галогена необходимы специальные условия. Если бромирование проводить в низкополярном растворителе (CCl4, CHCl3), образуются преимущественно монобромфенолы с преобладающим количеством пара-изомера. При хлорировании образуется преимущественно орто-изомер. Йод непосредственно не йодирует фенолы. Реакция нитрования фенола происходит при комнатной температуре уже при обработке разбавленной азотной кислотой, тогда как для нитрования бензола используют нитрующую смесь. При этом образуется смесь о- и п-нитрофенолов.
При действии концентрированной азотной кислоты фенол превращается в 2,4,6-тринитрофенол (пикриновую кислоту). Ранее пикриновую кислоту использовали как взрывчатое вещество.
Сульфирование фенола проходит очень легко и в зависимости от температурного режима приводит к образованию орто- или пара-изомеров:
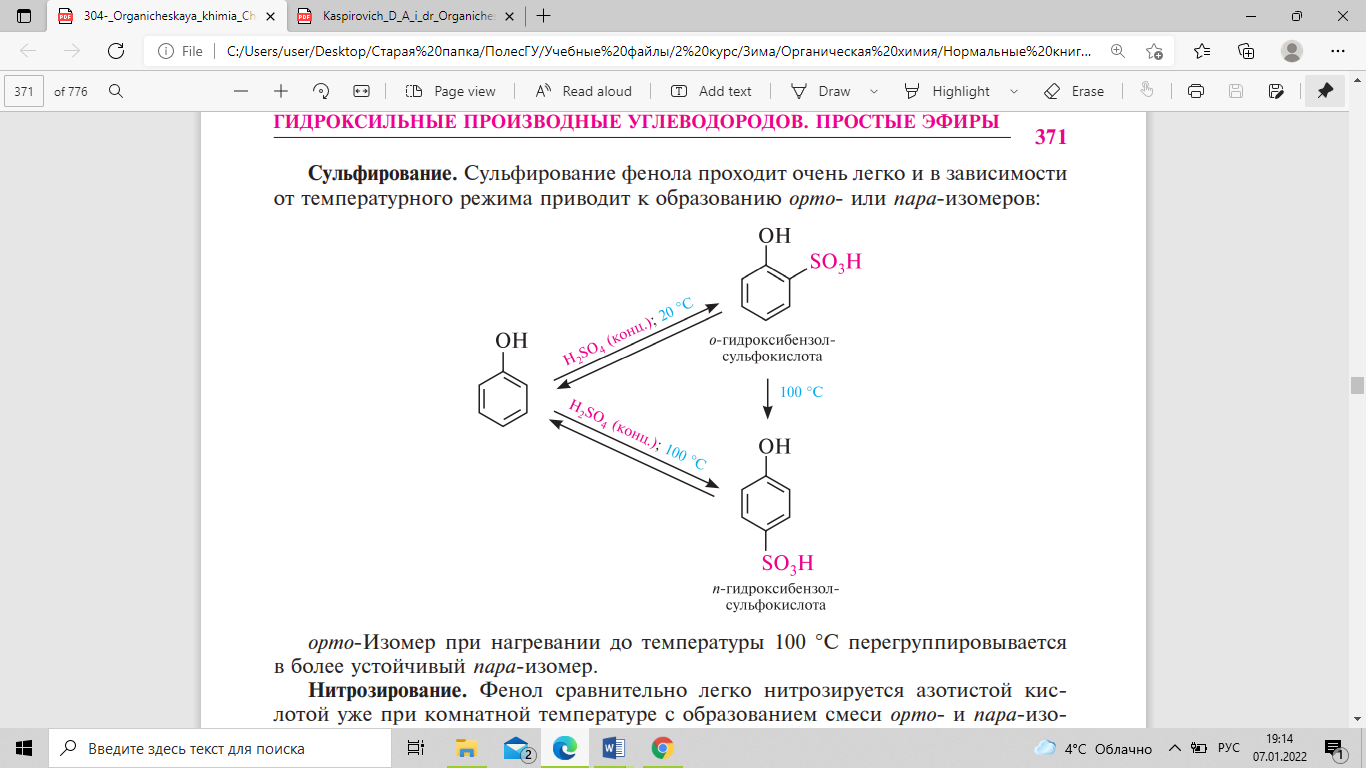
Орто-изомер при нагревании до температуры 100°с перегруппировывается в более устойчивый пара-изомер.
Алкилирование фенолов можно проводить по реакции фриделя—крафтса (с-алкилирование). Однако выходы в этой реакции, как правило, невысокие. Наиболее часто для алкилирования используют спирты и алкены в присутствии кислот (H2SO4, H3PO4 или BF3):

Карбоксилирование фенолятов щелочных металлов (реакция Кольбе). Окисление фенолов.
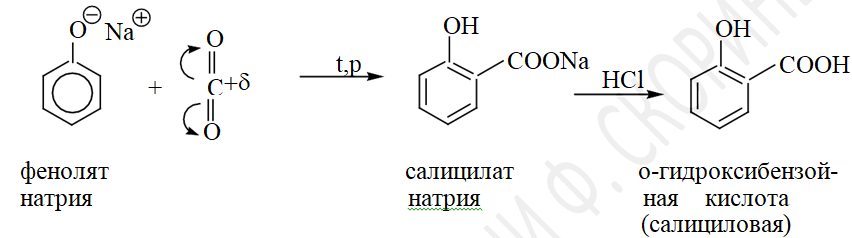
Карбоксилирование (синтез фенолокарбоновых кислот). При нагревании натрия фенолята в токе СО2 образуется натрия салицилат, который под действием минеральных кислот превращается в салициловую кислоту:
Окисление. Фенол и многие замещенные фенолы при хранении постепенно окисляются кислородом воздуха, образуя сложную смесь соединений. Если пара-положения бензольного кольца фенолов свободны, то одним из основных превращений феноксильных радикалов является димеризация по пара-положению с образованием 4,4'- дигидроксидифенила.
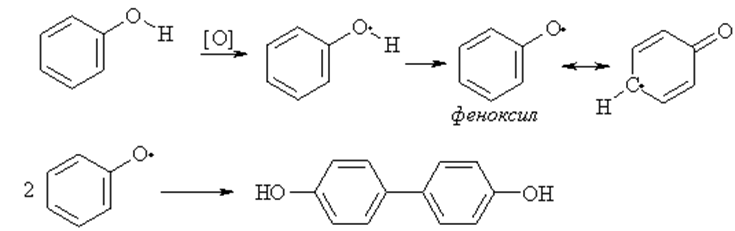
Феноксильные радикалы, имеющие в орто- и пара-положениях объемные заместители, настолько стабильны, что могут существовать в растворе длительное время и даже быть выделенными в кристаллическом виде.

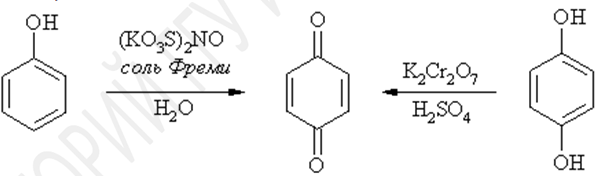
Фенол окисляется бихроматом калия или солью Фреми (нитрозодисульфонатом калия) до пара-бензохинона. При окислении двухатомных фенолов пирокатехина и гидрохинона, образуются орто- и пара-бензохиноны, соответственно.



Хиноны и их биологическая роль. Фенольные соединения в природе. Витамин Е. Флавоноиды.
Хиноны. Представляют собой циклические α,β-ненасыщенные дикетоны. Простейшими представителями этого класса соединений являются о-хинон и п-хинон:

Хиноны представляют собой окрашенные кристаллические вещества: о-хинон — ярко-красный, п-хинон — желтый.
Хиноны широко распространены в природе в качестве пигментов и биологически активных веществ, играющих важную роль в окислительно-восстановительных процессах, протекающих в живых организмах. Так, хиноидная структура входит в состав витаминов группы К, отвечающих за свертываемость крови, и убихинонов (коферментов Q), основная функция которых заключается в переносе электронов и протонов от различных субстратов к цитохромам при дыхании и окислительном фосфорилировании.
Фенольные соединения — вещества ароматической природы, содержащие одну или несколько гидроксильных групп. Фенольные соединения находятся в растениях, плодах и овощах преимущественно в виде гликозидов и реже в свободном виде Фенольные соединения являются основными антиоксидантами растительного происхождения, а их биологическая активность зависит от строения, количества, местонахождения гидроксильных групп в бензольном ядре и от степени полимеризации.
Флавоноиды — основная группа природных полифенолов. Флавоноиды играют важную роль в растительном метаболизме и очень широко распространены в высших растениях. Многие флавоноиды — пигменты, придающие разнообразную окраску растительным тканям. Так, антоцианы определяют красную, синюю, фиолетовую окраску цветов, а флавоны, флавонолы, ауроны, халконы — жёлтую и оранжевую. Они принимают участие в фотосинтезе, образовании лигнина и суберина, в качестве защитных агентов в патогенезе растений, вовлечены в регуляцию процессов прорастания семян, а также пролиферации и отмирания (путём апоптоза) клеток удлиняющихся растущих частей растений.
Витами́н E — группа природных соединений производных токола. Важнейшими соединениями являются токоферолы и токотриенолы. Жирорастворим. Витамин Е является универсальным протектором клеточных мембран от окислительного повреждения. Он занимает такое положение в мембране, которое препятствует контакту кислорода с ненасыщенными липидами мембран. Токоферол является не только антиоксидантом, но и антигипоксантом (группа лекарственных средств, улучшающих утилизацию циркулирующего в организме кислорода и повышающих устойчивость к гипоксии (кислородной недостаточности), что объясняется его способностью стабилизировать митохондриальную мембрану и экономить потребление кислорода клетками. Витамин контролирует биосинтез убихинона — компонента дыхательной цепи и главного антиоксиданта митохондрий
Флавоноиды, раститительные пигменты, представляющие собой гликозиды фенольного характера, содержащие в качестве агликона производные флавана (2-фенилхромана, ф-ла I). Разнообразие флавоноидов достигается вследствие того, что они, как правило, содержат в агликоне несколько гидроксильных или метоксильных групп. Все флаваноноиды - бесцветные кристаллы, легко образуют комплексы с ионами металлов, что используют для их идентификации методами спектрофотометрии. Антоцианы образуют также комплексы с флавоноидами в результате возникновения водородных связей между гидроксильными группами ангидрооснования антоцианидинов и гидроксильными группами в ароматическом ядре (копигментация). В результате такого взаимодействия достигается огромное разнообразие окраски цветков растений. Они защищают фотосинтезирующий аппарат клетки растений от повреждающего воздействия коротковолнового УФ излучения, обладают антимутагенной активностью и играют роль индукторов (сигнальных в-в) во взаимоотношениях растений с микроорганизмами. В ряде случаев флавоноиды служат защитными агентами при поражении растений патогенами.
Простые эфиры. Номенклатура, классификация. Расщепление кислотами. Образование гидропероксидов, их обнаружение и разложение. Циклические простые эфиры. Тетрагидрофуран. 1,4-Диоксан.
Простыми эфирами называют органические соединения общей формулы R–O–R'. Их также можно рассматривать как производные спиртов, енолов и фенолов, образующихся в результате замещения атома водорода гидроксильной группы углеводородным остатком. Радикалы в простых эфирах могут быть одинаковыми (симметричные эфиры) или разными (несимметричные, или смешанные эфиры). Простые эфиры могут иметь и циклическое строение. В циклических эфирах один или более атомов кислорода входят в состав цикла. Макроциклические полиэфиры общей формулы, где n = 4…20, называют краун-эфирами.
Названия простых эфиров по радикало-функциональной номенклатуре обычно образуют из названий углеводородных радикалов R и R´ (в алфавитном порядке), суффикса -овый и слова эфир. В заместительной номенклатуре IUPAC простые эфиры рассматривают как производные углеводородов, в которых один из атомов водорода замещен R-оксигруппой (RO—). За родоначальную структуру принимается более сложный по структуре радикал:
Циклические эфиры чаще рассматривают как гетероциклические соединения и для составления их названий применяют тривиальную и систематическую номенклатуру. В названиях краун-эфиров цифра в квадратных скобках указывает общее количество атомов в макроцикле, а другая цифра — количество атомов кислорода.
Изомерия простых эфиров обусловлена изомерией радикалов, связанных с атомом кислорода.
В химическом отношении простые эфиры являются весьма инертными веществами. они не реагируют с разведенными минеральными кислотами и щелочами на холоде. Простые эфиры проявляют слабые основные свойства за счет наличия на атоме кислорода неподеленных пар электронов. Они подвергаются расщеплению под действием йодоводородной и концентрированной серной кислот. Реакционная способность виниловых и ариловых эфиров обусловлена природой углеводородного радикала. Реакцию расщепления органических соединений под действием кислот называют ацидолиз. Концентрированные кислоты (HI, HBr, H2SO4) уже при комнатной температуре расщепляют простые эфиры. В реакции с йодоводородной кислотой при эквимолярном соотношении реагентов образуются галогенуглеводород и спирт, в избытке кислоты — только галогенуглеводороды.
В несимметричных эфирах нуклеофильной атаке преимущественно подвергается наиболее стерически доступный радикал. Реакция протекает по механизму SN2. При взаимодействии эквимолярных количеств галогеноводородной кислоты и диалкиловых эфиров, в составе которых один радикал первичный, а другой — третичный, аллильный или бензильный, образуется первичный спирт. Реакция протекает по механизму SN1. Расщепление простых эфиров под действием концентрированной серной кислоты приводит к образованию сложных эфиров серной кислоты.
Простые эфиры окисляются под действием кислорода воздуха с образованием взрывоопасных гидропероксидов R—O—он и пероксидов R—O—O—R. Поэтому перегонку эфиров нельзя вести досуха из-за опасности взрыва. для разрушения пероксидов эфир обрабатывают восстановителем, разрушающим пероксид. Хранят свободные от пероксидов эфиры над металлическим натрием или кальция гидридом.
Кроме свободно-радикального пути расщепления, алкилароматические гидропероксиды способны к распаду под влиянием кислотных и щелочных катализаторов. В присутствии уже небольшого количества сильной кислоты (например, 0,1 % H2SO4) гидропероксиды распадаются с образованием фенолов и карбонильных соединений.
Тетрагидрофуран — органическое химическое вещество, циклический простой эфир. Бесцветная легколетучая жидкость с характерным «эфирным» запахом. Важный апротонный растворитель. Широко применяется в органическом синтезе. Химическая формула: C4H8O. Используют как растворитель, например для поливинилхлорида, в лабораторной практике — вместо этилового эфира при получении магнийорганических соединений (в частности, винилмагнийбромида). Продукты гомо- и сополимеризации тетрагидрофурана — сырьё для получения уретановых каучуков. Из ТГФ синтезируют гамма-бутиролактон (по этой причине ТГФ включён в Таблицу III списка IV прекурсоров). Кроме того, используется как противокристаллизационная присадка к авиационным и ракетным топливам.
Диоксан — циклическое химическое соединение с формулой C4H8O2. Традиционно под диоксаном понимается 1,4-диоксан (диэтилендиоксид), циклический простой эфир. Диоксан применяется в органической химии в качестве полярного апротонного растворителя. В основном используется как стабилизатор 1,1,1-трихлорэтана. Имеет сладковатый запах, схожий с запахом диэтилового эфира. Диоксан — побочный продукт этоксилирования в производстве поверхностно-активных веществ, например, натрий лауретсульфата, этоксилированных спиртов и др.
Оксираны: получение, взаимодействие с водой, аммиаком и аминами, магнийорганическими соединениями. Краун-эфиры: комплексообразование с ионами металлов, применение.
Оксиранами (эпоксиды) называют трехчленные циклические соединения, содержащие один атом кислорода в цикле. Оксираны обладают высокой реакционной способностью в реакциях раскрытия трехчленного цикла под действием различных нуклеофильных агентов. Алкены окисляются перкислотами в неполярной среде с образованием эпоксидов. Простейший и наиболее важный из оксиранов - окись этилена получается в промышленности каталитическим окислением этилена.
В отличии от простых эфиров для оксиранов характерны реакции расщепления напряженного трехчленного цикла под действием разнообразных нуклеофильных агентов. При наличии в оксирановом кольце алкильного или арильного заместителя атака нуклеофильного агента направляется преимущественно по незамещенному, пространственно более доступному атому углерода.
Механизм гидролиза и алкоголиза оксиранов может полностью измениться при проведении реакции в кислой среде или в присутствии электрофильного катализатора.
В быстрой и обратимой первой стадии происходит протонирование оксирана по атому кислорода с образованием оксониевого катиона.
Во второй медленной стадии протонированная форма подвергается нуклеофильной атаке водой, спиртом или галогенид-ионом. Протонирование оксирана ускоряет раскрытие кольца при взаимодействии с нуклеофильными агентами. В зависимости от строения оксирана механизм расщепления трехчленного цикла может изменяться от SN2 до SN1. Если в результате раскрытия цикла образуется относительно устойчивый третичный карбокатион, катализируемый кислотой сольволиз оксирана протекает по SN1-механизму. Направление раскрытия оксиранового кольца при SN1-механизме полностью противоположно тому, которое наблюдается при SN2-механизме. Если реализуется SN1-механизм, нуклеофильная атака направляется по наиболее замещенному атому углерода. При конкуренции SN2 и SN1-механизмов раскрытие цикла не отличается высокой региоселективностью и приводит к образованию смеси двух изомерных продуктов сольволитического расщепления кольца оксирана.
Взаимодействие оксиранов с магнийорганическими соединениями следует рассматривать как бимолекулярное нуклеофильное замещение у насыщенного атома углерода оксирана под действием карбоаниона металлоорганического соединения. Раскрытию цикла способствует образование координационого донорно-акцепторного комплекса между атомами кислорода и магния, которое можно рассматривать как электрофильный катализ в реакции нуклеофильного замещения. Как следует из приведенного уравнения продуктами этой реакции являются спирты. Оксирановый цикл размыкается также под действием диалкилкупратов, причем замещение происходит при наименее замещенном атоме углерода. Краун-эфиры (краун-соединения), макрогетероциклические полиэфиры, содержащие несколько атомов кислорода в цикле, в названиях краун-эфиров первая цифра указывает на размер цикла, а вторая определяет число атомов кислорода в цикле. Краун-эфиры образуют макроциклические комплексные соединения как с катионами, так и с нейтральными молекулами. В комплексах краун-эфира с катионами металлов (щелочных, щёлочноземельных и др.) катион включается во внутримолекулярную полость краун-эфира и удерживается там благодаря ион-дипольному взаимодействию с гетероатомами .Селективность связывания и устойчивость образуемых краун-эфиром комплексов зависит от ряда факторов – геометрического соответствия размеров внедряющейся частицы и полости макроцикла, природы растворителя, хелатного и макроциклических эффектов, заряда внедряющейся частицы, природы противоиона и пр. За счёт комплексообразования краун-эфиры повышают растворимость солей щелочных и щёлочноземельных металлов в малополярных растворителях; пример - образование малинового бензола и оранжевого бензола (окислительных реагентов) при растворении в бензоле соответственно KMnО4 или К2Cr2О7 в присутствии краун-эфира. Щелочные металлы в присутствии краун-эфира растворяются в координирующих органических растворителях; интенсивный синий цвет образующихся растворов объясняется присутствием в них сольватированной формы свободных электронов (получаемый раствор является восстановительным реагентом). Краун-эфиры способны селективно экстрагировать ионы металлов и некоторые органические соединения (амины, аминокислоты и др.) из водной фазы в органическую, энантиоспецифически связывать хиральные молекулы (в частности, протонированные формы аминокислот). За счёт влияния на ионную и субстратную проницаемость биологических мембран, на ферментные системы и другие биохимические процессы краун-эфиры проявляют различные виды биологической активности. Краун-эфир широко используют в научных исследованиях как доступные и разнообразные модели при изучении сложных природных супрамолекулярных систем. С помощью краун-эфира можно избирательно выводить из организма ионы тяжёлых металлов или вводить малые количества нужных ионов или молекул. Краун-эфиры применяют для концентрирования, разделения, очистки и регенерации металлов, в том числе лантаноидов, для разделения нуклидов, энантиомеров; для создания ионоселективных датчиков и мембран; в межфазном катализе и для ускорения реакций, проходящих с участием анионов; как лекарственные препараты, пестициды, антидоты.
Кислотность тиолов. Нуклеофильные свойства тиолов, тиолятов и органических сульфидов. Окисление тиолов. Образование дисульфидов и их роль в биохимических процессах.
Тиолами называют производные углеводородов, в молекулах которых один или несколько атомов водорода замещены меркаптогруппой —SH. Тиолы можно также рассматривать как тиоаналоги гидроксильных производных углеводородов, в молекулах которых атом кислорода группы –OH заменен на атом серы. Для соединений этого класса используют еще название «меркаптаны». Тиолы, в которых меркаптогруппа связана с алифатическим радикалом, называют тиоспиртами, а с ароматическим радикалом — тиофенолами.
По химическим свойствам тиолы во многом сходны с гидроксильными производными углеводородов. Особенности их химического поведения обусловлены уменьшением прочности связи S—Н по сравнению со связью O—H в спиртах и фенолах. Именно поэтому тиолы обладают более выраженными кислотными свойствами, чем соответствующие им гидроксианалоги. реакции тиолов в основном обусловлены ионизацией связи S—H и нуклеофильными свойствами атома серы. Тиолы, как более сильные кислоты, чем аналогичные OH-кислоты, легко образуют соли — тиоляты (меркаптиды) не только со щелочными металлами, но также и с ионами тяжелых металлов. На этом свойстве тиолов основано их применение в медицине в качестве антидотов при отравлении тяжелыми металлами. Реакция тиолов с карбоновыми кислотами катализируется сильными кислотами и приводит к образованию тиоэфиров карбоновых кислот. Данная реакция аналогична реакции этерификации карбоновых кислот и спиртов.
В отличие от спиртов, тиолы окисляются не по атому углерода, а по атому серы. Продукты реакции зависят от условий окисления. При окислении тиолов в мягких условиях (H2O2, CuCl2 и др.) образуются диалкилдисульфиды.
При окислении тиолов сильными окислителями (KMnO4, HNO3 или HOI) образуются сульфокислоты. Сульфиды (тиоэфиры) можно рассматривать как производные сероводорода Н2S, в молекуле которого два атома водорода замещены углеводородными радикалами. Алкилирование тиолятов. Этод метод аналогичен синтезу простых эфиров по Вильямсону.
Нуклеофильные свойства тиолов, тиолятов и органических сульфидов.
Тиолы и сульфиды проявляют высокое сродство к атому углерода с пониженной электронной плотностью. Они не образуют прочных водородных связей с протонными растворителями, и их неподеленные пары электронов доступны для взаимодействия с органическими реагентами. Эти две причины обусловливают высокую нуклеофильность тиолов, сульфидов.
Сульфиды по своим химическим свойствам во многом напоминают простые эфиры, однако для них характерны особенности, связанные с большей поляризуемостью атома серы по сравнению с атомом кислорода. Сравнительно легко реагируют с галогеналканами с образованием достаточно стабильных галогенидов триалкилсульфония. Сульфиды могут быть также получены в результате прямого взаимодействия сульфида натрия с двумя молями алкилирующего агента. Высокая нуклеофильность атома серы в сульфидах открывает возможность для получения солей сульфония в результате алкилирования сульфидов.
Окисление тиолов резко отличается от окисления спиртов. В зависимости от природы окислителя продуктами окисления тиолов являются дисульфиды R-S-S-R, сульфиновые RSO2H или сульфоновые RSO3H кислоты. При действии таких окислителей, как йод, бром, пероксид водорода, МпО3, тиолы окисляются до дисульфидов Дисульфиды легко восстанавливаются обратно до тиолов цинком в уксусной кислоте или лучше всего раствором щелочного металла в жидком аммиаке. Сильные окислители - азотная кислота или перманганат калия - окисляют тиолы до сульфоновых кислот (продуктов исчерпывающего окисления органических соединений серы). Дисульфиды используют для получения пестицидов, медпрепаратов, красителей, в органическом синтезе. Дисульфидные связи содержатся в молекулах многих белков, некоторых природных низкомолекулярных биологически активных соединениях (напр., в цистине, липоевой к-те). К природным дисульфидам относятся спородесмины и некоторые антибиотики. Дисульфиды средне- или малотоксичны для теплокровных.
Классификация, номенклатура и изомерия аминов. Алифатические и ароматические амины, первичные, вторичные и третичные амины.
Аминами называют производные аммиака, в молекуле которого один, два или три атома водорода замещены углеводородными радикалами. Соответственно числу углеводородных остатков различают первичные, вторичные и третичные амины. В зависимости от природы углеводородных радикалов у атома азота амины подразделяют на алифатические, алициклические и ароматические (аренамины); амины, у которых атом азота связан с алифатическим и ароматическим углеводородным радикалом, называют смешанными:

По заместительной номенклатуре IUPAC названия первичных аминов образуют путем добавления к названию углеводорода суффикса -амин, указывая положение аминогруппы в углеродной цепи. При составлении названий вторичных и третичных аминов их рассматривают как производные первичного амина с заместителями при атоме азота. За исходный первичный амин в этом случае принимается связанный с атомом азота наиболее сложный по структуре радикал. Остальные углеводородные заместители при атоме азота перечисляют в алфавитном порядке с указанием локанта N-.
Если соединение содержит две или три аминогруппы, то в названии их обозначают множительными приставками ди- или три-, которые ставятся перед суффиксом –амин. Простейшие амины чаще всего называют по радикало-функциональной номенклатуре. Согласно этой номенклатуре, названия аминов образуют из названий углеводородных радикалов, перечисляемых в алфавитном порядке, и суффикса -амин. Некоторые амины сохраняют тривиальные названия. Названия первичных ароматических аминов, а также смешанных аминов обычно образуют на основе названия родоначального представителя — анилина. В случае смешанных аминов положение заместителей у атома азота обозначают с помощью локанта N-:
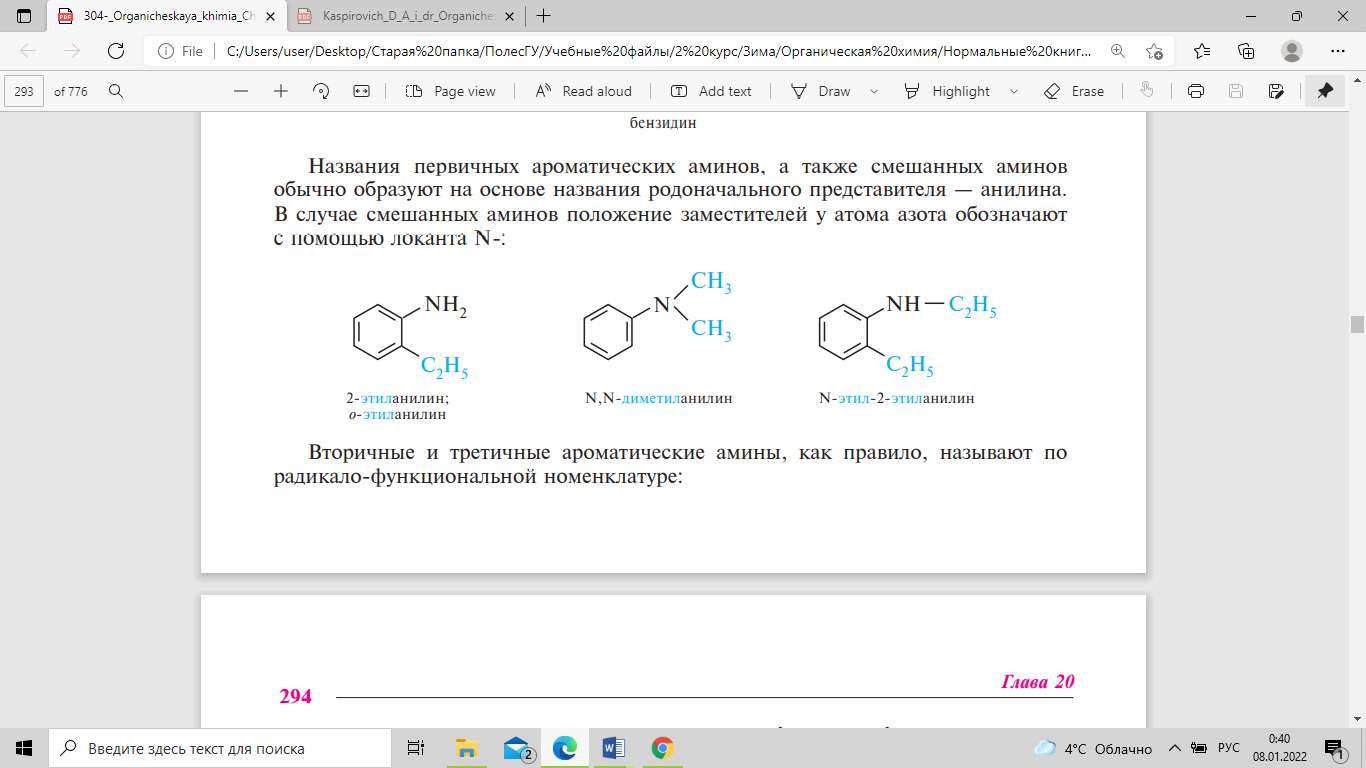
Вторичные и третичные ароматические амины, как правило, называют по радикало-функциональной номенклатуре.
Изомерия аминов обусловлена разной структурой углеводородных радикалов, разным положением аминогруппы и метамерией. Сущность метамерии состоит в том, что амины с одной и той же брутто-формулой могут быть первичными, вторичными и третичными. Приведенные соединения являются метамерами:

Электронное строение аминов. Роль неподеленной электронной пары азота в проявлении основных и нуклеофильных свойств алкил- и ариламинов. Реакции ацилирования и алкилирования аминов. Аммониевые соли.
Электронное строение аминов:
Атом азота в аминах находится в состоянии sp3-гибридизации и имеет тетраэдрическую ориентацию орбиталей в пространстве.
Три из четырёх гибридных орбиталей участвуют в образовании σ-связей N–C или N–H.
На четвёртой орбитали находится неподелённая электронная пара, которая обусловливает основные свойства аминов.
Два неспаренных электрона в молекулах аминов могут участвовать в образовании химической связи по донорно-акцепторному механизму.
Основные свойства алкил- и ариламинов обусловлены способностью атома азота с неподеленной электронной парой к присоединению протона с образованием иона замещённого аммония.
Алкильные радикалы увеличивают электронную плотность на атоме азота за счёт проявления электронодонорного эффекта (+I-эффекта), что приводит к заметному усилению основности.
Нуклеофильные свойства аминов также обусловлены наличием неподеленной пары электронов атома азота. Они проявляются в реакциях алкилирования аминов, взаимодействия с карбонильными соединениями и ацилирования производными карбоновых кислот.
Ароматические амины менее сильные основания, чем аммиак и алкиламины. Делокализация неподеленной электронной пары по бензольному кольцу уменьшает электронную плотность на атоме азота, и, следовательно, понижается способность к связыванию протона.
Реакция алкилирования аминов— это реакция нуклеофильного замещения, в которой амины подвергаются алкилированию галогеналканами. В результате алкилирования аммиака образуется первичный амин, из первичных аминов — вторичные, из вторичных — третичные, из третичных — четвертичные аммониевые соли.
Реакция ацилирования аминов — это реакция первичных и вторичных аминов с функциональными производными карбоновых кислот: галогенангидридами, ангидридами и сложными эфирами. В результате ацилирования аминов образуются N-замещённые амиды карбоновых кислот. Третичные амины не содержат при атоме азота водорода и поэтому в реакцию ацилирования не вступают.
Соли аммония — сложные вещества, образованные катионом аммония NH4+ и кислотным остатком. По свойствам похожи на соли натрия или калия. Они имеют ионное строение и представляют собой твёрдые белые вещества, хорошо растворяющиеся в воде.
Примеры солей аммония:
NH4Cl — хлорид аммония (нашатырь);
(NH4)2SO4 — сульфат аммония;
NH4NO3 — нитрат аммония (аммиачная селитра).
Получаются соли аммония при взаимодействии аммиака или гидроксида аммония с кислотами.
Применяются соли аммония в качестве удобрений, в пищевой и текстильной промышленности
Особенности свойств ариламинов. Реакции электрофильного замещения в бензольном ядре ариламинов и их производных. Реакции диазотирования, соли арилдиазония. Реакции солей арилдиазония с выделением азота (замещение диазогруппы) и без выделения азота (азосочетание). Азокрасители.
Ариламинами называют производные аммиака, в молекуле которого один, два или три атома водорода замещены остатками ароматических углеводородов. Для ариламинов характерны реакции с участием аминогруппы и реакции с участием атомов углерода ароматического ядра.
За счет наличия неподеленной пары электронов на атоме азота проявляют основные свойства. Однако основность ариламинов значительно ниже основности алкиламинов, что обусловлено сопряжением неподелённой пары электронов атома азота с π-электронной системой ароматического ядра. В результате сопряжения неподеленная пара электронов частично делокализуется по ароматическому ядру, и поэтому она становится менее доступной для координации с протоном. На основность ариламинов существенное влияние оказывают заместители в бензольном кольце. Электронодонорные заместители увеличивают основность, а электроноакцепторные — уменьшают ее. Основность сильно снижается при переходе от первичных к третичным. Ариламины образуют соли только с сильными минеральными кислотами. Для ариламинов характерны реакции электрофильного замещения по ароматическому ядру, свойственные ароматическим углеводородам. Аминогруппа в молекуле ариламина в результате проявления +М-эффекта выступает в качестве сильного электронодонорного заместителя по отношению к бензольному кольцу и тем самым активизирует его реакционную способность в реакциях электрофильного замещения. Поэтому ариламины вступают в реакции электрофильного замещения значительно легче, чем бензол. Аминогруппа, являясь заместителем I рода, направляет электрофильное замещение в орто- и пара-положения.
Анилин легко реагирует с галогенами (Cl2, Br2) в отсутствие катализатора, образуя 2,4,6-тригалогенопроизводные ариламины. Так, при обработке анилина бромной водой практически с количественным выходом образуется осадок 2,4,6-триброманилина:

Хлор в водных растворах окисляет анилин, поэтому хлорирование с образованием 2,4,6-трихлоранилина осуществляют действием хлора в неводных растворителях. При получении моногалогенозамещенных ариламинов амины первоначально переводят в N-ацильные производные, которые затем галогенируют и гидролизуют. N-Ацетиламиногруппа является заместителем I рода, но ее активирующее влияние на бензольное кольцо значительно меньше, чем аминогруппы, так как неподеленная пара электронов атома азота участвует в сопряжении не только с π-электронной системой бензольного ядра, но и с π-электронами двойной связи карбонильной группы.
Нитрование ариламинов в отличие от аренов имеет ряд особенностей. Прямое нитрование ароматических аминов концентрированной азотной кислотой осуществить невозможно, так как они легко окисляются. При использовании в качестве нитрующего реагента нитрующей смеси ариламины, наряду с частично протекающими окислительными процессами, превращаются в ариламмонийные соли. Аммонийная группа, являясь электроноакцепторным заместителем, затрудняет нитрование и способствует образованию преимущественно мета-изомера. С целью защиты аминогруппы от процессов окисления и протонирования по атому азота ароматические амины предварительно ацетилируют. N-Ацетильные производные в отличие от аминов являются очень слабыми основаниями и даже в сильнокислой среде реагируют в непротонированной форме. Вместе с тем N-ацетиламиногруппа сохраняет электронодонорные свойства и ориентирует нитрование в орто- и пара-положения. Соотношение орто- и пара-изомеров зависит от состава нитрующей смеси и условий проведения реакции. После нитрования N-ацильные производные гидролизуют в кислой или щелочной среде. При нагревании анилина с концентрированной серной кислотой в среде высококипящего растворителя образуется n-аминобензолсульфокислота, которую чаще называют сульфаниловой кислотой. Реакция протекает через стадию образования N-фенилсульфаминовой кислоты, которая перегруппировывается в n-аминобензолсульфокислоту:

Сульфаниловая
кислота является структурным фрагментом
большой группы ЛП — сульфаниламидов.
Соли арендиазония образуются
при взаимодействии первичных ароматических
аминов с азотистой кислотой. В
промышленности соли арендиазония нашли
широкое применение для получения
разнообразных азокрасителей всех цветов
и оттенков. По этой причине диазотирование
относится к числу важнейших и наиболее
изученных реакций в органической
химии. Диазотирование первичных
ароматических аминов описывается
следующим суммарным уравнением: ArNH2+
NaNO2+
2 HCl![]() ArN+=N
Cl - +
NaCl + 2 H2O Реакции
с выделением азота.
При кипячении кислых растворов солей
диазония происходит выделение азота и
получаются фенолы. Превращение солей
диазония без выделения азота. Реакции
этой группы делают возможным переход
от диазосоединений к азосоединениям
(производным азобензола). Органические
вещества этого класса лежат в основе
одного из разделов промышленности,
производящей синтетические красители
из продуктов, добываемых из каменноугольного
дегтя. Все азокрасители получаются
при помощи так называемой реакции
сочетания солей диазония — химическая
реакция, в ходе которой к ароматическому
диазосоединению присоединяется другое
соединение, называемое азосоставляющим,
содержащее способный к замещению атом
водорода, либо в некоторых случаях,
другие атомы или группы, в результате
чего происходит образование азосоединения.
Диазосоединение при вступлении в реакцию
азосочетания находится в форме катиона
диазония. Сама реакция имеет две ступени,
из которых медленной является первая
— присоединение катиона к азосоставляющей.
Вторая стадия — отщепление протона,
которую можно ускорить при помощи
введения в реакционную массу акцепторов
протонов, в роли которых используют
карбонатные и ацетатные группы. Пример
реакции азосочетания — получение
4-гидроксиазобензола:
ArN+=N
Cl - +
NaCl + 2 H2O Реакции
с выделением азота.
При кипячении кислых растворов солей
диазония происходит выделение азота и
получаются фенолы. Превращение солей
диазония без выделения азота. Реакции
этой группы делают возможным переход
от диазосоединений к азосоединениям
(производным азобензола). Органические
вещества этого класса лежат в основе
одного из разделов промышленности,
производящей синтетические красители
из продуктов, добываемых из каменноугольного
дегтя. Все азокрасители получаются
при помощи так называемой реакции
сочетания солей диазония — химическая
реакция, в ходе которой к ароматическому
диазосоединению присоединяется другое
соединение, называемое азосоставляющим,
содержащее способный к замещению атом
водорода, либо в некоторых случаях,
другие атомы или группы, в результате
чего происходит образование азосоединения.
Диазосоединение при вступлении в реакцию
азосочетания находится в форме катиона
диазония. Сама реакция имеет две ступени,
из которых медленной является первая
— присоединение катиона к азосоставляющей.
Вторая стадия — отщепление протона,
которую можно ускорить при помощи
введения в реакционную массу акцепторов
протонов, в роли которых используют
карбонатные и ацетатные группы. Пример
реакции азосочетания — получение
4-гидроксиазобензола:  Реакцию
азосочетания обычно проводят при
температуре от 0 до 25 °C, повышая её при
необходимости до 40—50 °C в случаях
малоактивных диазосоединений, так как
при этом последние разрушаются с
выделением азота.
Реакцию
азосочетания обычно проводят при
температуре от 0 до 25 °C, повышая её при
необходимости до 40—50 °C в случаях
малоактивных диазосоединений, так как
при этом последние разрушаются с
выделением азота.
Классификация, номенклатура и изомерия карбонильных соединений.
Альдегидами и кетонами называют производные углеводородов, содержащие карбонильную группу С=О. В молекулах альдегидов карбонильная группа связана с атомом водорода и углеводородным радикалом. Группировку называют альдегидной группой. В молекулах кетонов карбонильная группа связана с двумя углеводородными радикалами и называется кетогруппой. Альдегиды и кетоны относят к группе карбонильных соединений. В зависимости от строения углеводородного радикала альдегиды и кетоны подразделяют на алифатические, алициклические и ароматические. среди алифатических альдегидов и кетонов различают насыщенные и ненасыщенные. В номенклатуре альдегидов и кетонов используют тривиальные и систематические названия. Тривиальные названия альдегидов происходят от названия кислот, в которые они превращаются при окислении. Альдегид, при окислении которого получают муравьиную кислоту, называют муравьиный альдегид или формальдегид (от лат. acidum formicum); альдегид, при окислении которого образуется уксусная кислота,— уксусный альдегид или ацетальдегид (от лат. acidum aceticum) и т. д. По заместительной номенклатуре IUPAC названия альдегидов образуют от названия углеводорода с тем же числом атомов углерода (включая атом углерода альдегидной группы), прибавляя суффикс -аль. нумерацию главной углеродной цепи начинают с атома углерода альдегидной группы. Нередко в названиях альдегидов положение заместителей указывают греческими буквами α, β, γ и др. (буквой α обозначают атом углерода, соседний с альдегидной группой):

Для названий кетонов широко используют радикало-функциональную номенклатуру, согласно которой к названиям (в алфавитном порядке) углеводородных радикалов при карбонильной группе прибавляют суффикс -кетон: При составлении названия кетонов по заместительной номенклатуре выбирают самую длинную углеродную цепь, в состав которой входит кетогруппа. Нумерацию проводят таким образом, чтобы атом углерода карбонильной группы получил возможно меньший номер. затем к названию предельного углеводорода, содержащего такое же количество атомов углерода, прибавляют суффикс -он и цифрой обозначают атом углерода, входящий в кетогруппу:

Для некоторых кетонов сохранились тривиальные названия - диметилкетон чаще называют ацетоном. Альдегиды и кетоны, содержащие одинаковое количество атомов углерода, изомерны между собой. так, пропанон и пропаналь являются структурными изомерами (изомерами функциональной группы). Изомерия альдегидов и кетонов может быть связана с различной структурой углеродной цепи:

Лля кетонов характерна также изомерия, обусловленная положением карбонильной группы:

Строение карбонильной группы в альдегидах и кетонах и Реакции нуклеофильного присоединения (реактивами Гриньяра, циановодородом). Механизм реакций.
Карбонильная группа в альдегидах и кетонах — это двойная связь между атомами углерода и кислорода, которая сильно поляризована. В силу значительной полярности связи C=О она склонна к гетеролитическому разрыву, а атом углерода имеет электрофильный характер и восприимчив к атаке нуклеофильных реагентов.
Реакции нуклеофильного присоединения к карбонильной группе:
Присоединение воды. Образующиеся гем-диолы неустойчивы, и равновесие в этой реакции сильно смещено влево.
Присоединение цианидов. Реакция катализируется цианистым калием или натрием. Образующиеся оксинитрилы (или циангидрины) могут быть гидролизованы до оксикарбоновых кислот.
Присоединение спиртов. При присоединении первой молекулы спирта образуются полуацетали. Реакция катализируется кислотами или основаниями.
Присоединение реактивов Гриньяра. Взаимодействие металлорганических соединений типа R-Mg-X с карбонильными группами приводит к первичным, вторичным и третичным спиртам, соответственно.
Присоединение аммиака и аминов. Первичные амины присоединяются к альдегидам и кетонам с образованием иминов. Аналогичная реакция вторичных аминов с карбонильными соединениями даёт енамины.
Реакции карбонильных соединений с гетеронуклеофилами: присоединение воды и спиртов, образование ацеталей. Реакции карбонильных соединений с аммиаком, аминами.
Растворение альдегидов в воде сопровождается образованием гидратов, которые представляют собой продукты присоединения молекулы воды по карбонильной группе. как правило, гидраты альдегидов неустойчивы. В водных растворах они находятся в динамическом равновесии с альдегидом:

Положение равновесия определяется строением карбонильных соединений. Формальдегид в воде практически полностью гидратирован, ацетальдегид — наполовину, а ацетон — почти не взаимодействует с водой. Гидраты альдегидов существуют только в растворе и выделить их невозможно; при перегонке они разлагаются. Существование гидратов доказывают с помощью физических методов исследования. В некоторых случаях, когда карбонильная группа связана с сильным электроноакцепторным заместителем, образовавшийся гидрат может быть выделен в свободном виде. трихлорацетальдегид (хлораль), присоединяя молекулу воды, превращается в хлоралгидрат, представляющий собой устойчивое кристаллическое вещество. Отщепить воду от хлоралгидрата удается только при действии серной кислоты. Хлоралгидрат применяется в медицинской практике как успокаивающее и противосудорожное средство. При взаимодействии альдегидов со спиртами образуются полуацетали, а в присутствии следов минеральных кислот — ацетали. Полуацетали, как правило, малоустойчивы. исключение составляют циклические полуацетали, образующиеся самопроизвольно из γ- и δ-гидроксиальдегидов:
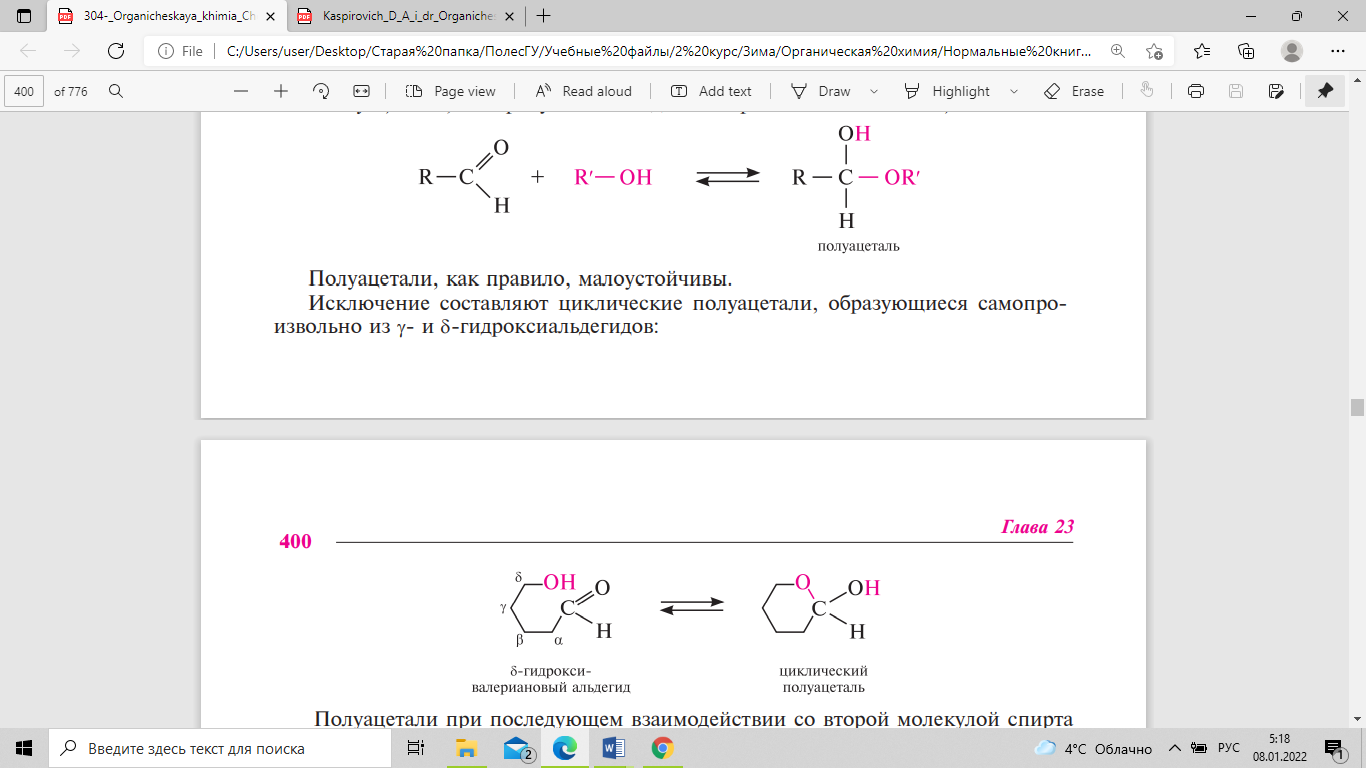
Полуацетали при последующем взаимодействии со второй молекулой спирта превращаются в ацетали. Ацетали устойчивы в щелочной среде, но легко гидролизуются до свободного альдегида в разбавленных кислотах. такое свойство ацеталей используется в органическом синтезе для защиты альдегидной группы. Кетоны из-за низкой реакционной способности и пространственных препятствий со спиртами не взаимодействуют, поэтому кетали получают, используя другие синтетические методы. Альдегиды, присоединяя молекулу аммиака, образуют альдимины. в процессе реакции вначале образуется неустойчивый аминоспирт, от которого затем отщепляется молекула воды. По аналогичному механизму протекают реакции с другими азотистыми основаниями. Альдимины — неустойчивые соединения, они самопроизвольно циклотримеризуются с образованием альдегидаммиака:

Кетоны также взаимодействуют с аммиаком, но при этом образуются продукты более сложного строения. Альдегиды и кетоны реагируют с первичными аминами с образованием N-замещенных иминов (азометинов). Кетоны реагируют медленнее, чем альдегиды. N-замещенные имины, содержащие в структуре хотя бы один арильный радикал, отличаются значительной устойчивостью; их называют основаниями Шиффа.

Реакции енольных форм карбонильных соединений: - галогенирование, галоформное расщепление, изотопный обмен водорода. Альдольно-кротоновая конденсация, кислотный и основный катализ.
Реакция галогенирования. Альдегиды и кетоны как сн-кислоты легко вступают в реакции с галогенами с образованием -галогенозамещенных продуктов:


Для метилкетонов и ацетальдегида характерна галоформная реакция. При взаимодействии с хлором, бромом или йодом в щелочной среде они галогенируются по метильной группе. Полученные продукты — тригалогенкетон или тригалогенацетальдегид — расщепляются в щелочной среде на соль карбоновой кислоты и галоформ (хлороформ, бромоформ, йодоформ). в случае йодирования в щелочной среде реакция идет с выделением йодоформа снІ3 — кристаллического вещества желтого цвета c характерным запахом:
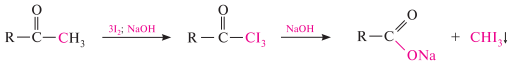
Альдольная конденсация. альдегиды, содержащие атомы водорода у -углеродного атома, в присутствии каталитических количеств основания способны вступать в реакцию альдольной конденсации. взаимодействие осуществляется при участии подвижного -атома водорода одной молекулы и карбонильной группы другой молекулы альдегида и приводит к образованию соединения со спиртовой и альдегидной группой (альдоль).

Переход от предельных альдегидов через стадию образования альдоля с последующей внутримолекулярной дегидратацией к ,-непредельным альдегидам называют кротоновой конденсацией.

Взаимодействие неенолизирующихся альдегидов со щелочами (реакция Канниццаро). Реакции окисления и восстановления карбонильных соединений.
Реакция Канниццаро — это окислительно-восстановительное превращение (диспропорционирование) неенолизируемых альдегидов, не содержащих атомов водорода у α-углеродного атома, при действии щелочи. В результате реакции половина молекул альдегида восстанавливается в спирт, а другая окисляется до карбоновой кислоты.
Примеры альдегидов, которые могут подвергнуться реакции Канниццаро: ароматические альдегиды, формальдегид, а также альдегиды, у которых карбонильная группа связана с третичным углеродным атомом.
Реакция Канниццаро открыта в 1853 году, в присутствии сильного основания или концентрированной щелочи ароматические альдегиды вступают в реакцию диспропорционирования. При этом одна из двух молекул альдегида окисляется до соответствующей кислоты, а другая — восстанавливается до спирта:
![]()
Реакция протекает с переносом гидридного иона (Н–). В реакцию канниццаро вступают также некоторые альдегиды жирного ряда, в частности формальдегид, и альдегиды, не содержащие атомов водорода при альфа-углеродном атоме. Альдегиды, содержащие атомы водорода при альфа-углеродном атоме, в условиях реакции канниццаро осмоляются. Реакцию восстановления альдегидов и кетонов широко используют для получения спиртов (альдегиды восстанавливаются до первичных, а кетоны — до вторичных спиртов). в технике спирты получают в результате каталитического гидрирования; присоединение водорода происходит в присутствии кобальта, никеля или платины:

Альдегиды и кетоны по-разному относятся к действию окислителей. альдегиды очень легко окисляются; даже при действии таких слабых окислителей, какими являются ионы Ag+ и сu2+, они превращаются в карбоновые кислоты. Реакцию окисления альдегидов аммиачным раствором серебра оксида (реактив Толленса) называют реакцией «серебряного зеркала». Ион серебра в этой реакции восстанавливается до свободного серебра, которое выделяется в виде зеркала на стенках пробирки: Алифатические альдегиды также восстанавливают реактив Фелинга (смесь раствора меди (ІІ) сульфата со щелочным раствором калий-натриевой соли виннокаменной кислоты). При нагревании альдегидов с фелинговой жидкостью образуется кирпично-красный осадок меди (I) оксида. Реакции окисления альдегидов аммиачным раствором серебра оксида и реактивом Фелинга используются в аналитической практике для обнаружения альдегидной группы. Кетоны в этих условиях не окисляются, поэтому эти реакции могут быть использованы и для отличия альдегидов от кетонов. Окисление кетонов происходит только в присутствии сильных окислителей (KMnO4, K2сr2O7 и др.). При этом происходит разрыв связей с—с между атомами углерода карбонильной группы и углеводородного радикала. В результате реакции образуется смесь карбоновых кислот:

По продуктам реакции окисления кетонов определяют их строение.
Реакции окисления карбонильных соединений:
Окисление альдегидов слабыми окислителями сопровождается образованием карбоновых кислот с таким же, как в исходном альдегиде, числом углеродных атомов. Примеры реакций: R-СН=О + Аg(NН3)2ОН → R-СООН + Аg + NН4ОН + NН3 (реакция «серебряного зеркала»), R-СН=О + Cu (ОН)2 → R-СООН + Cu2О + Н2О.
Кетоны в этих условиях не окисляются, окисление с разрывом углерод-углеродных связей протекает довольно в жёстких условиях с образованием соединений (кетоны, карбоновые кислоты), содержащих по сравнению с исходным, меньшее число атомов углерода.
Реакции восстановления карбонильных соединений:
При восстановлении альдегидов образуются первичные спирты, кетонов — вторичные спирты.
Под действием концентрированной щелочи альдегиды, у которых отсутствует атом водорода у α-углеродного атома, могут восстанавливаться до углеводородов.
Монокарбоновые кислоты. Номенклатура. Строение карбоксильной группы и карбоксилат-иона. Кислотность карбоновых кислот.
Одноосновные (монокарбоновые) насыщенные карбоновые кислоты — органические соединения, состоящие из насыщенного углеводородного заместителя и одной карбоксильной группы −COOH.
Примеры монокарбоновых кислот:
НСООН — муравьиная кислота, метановая кислота.
СН3СООН — уксусная, этановая кислота.
СН3СН2СООН — пропионовая, пропановая кислота.
СН3СН2СН2СООН — масляная, бутановая кислота.
СН3СН(СН3)СООН — изомасляная или 2-метилпропановая кислота.
СН3СН2СН(СН3) СООН — α-метилмасляная, 2-метилбутановая кислота.
СН3(СН2)3СООН — валериановая, пентановая кислота.
СН3(СН2)4СООН — капроновая, гексановая кислота.
Номенклатура монокарбоновых кислот формируется путём добавления суффикса «овая» к названию алкана с соответствующим числом атомов углерода и слова «кислота».
Примеры:
Метановая кислота — HCOOH — муравьиная кислота.
Этановая кислота — CH3-COOH — уксусная кислота.
Пропановая кислота — C2H5-COOH — пропионовая кислота.
Бутановая кислота — C3H7-COOH — масляная кислота.
Пентановая кислота — C4H9-COOH — валериановая кислота.
Многие монокарбоновые кислоты имеют также тривиальные названия.
Карбоксильная группа (карбоксил, карбоксигруппа) –СООН состоит из двух функциональных групп — карбоксильной группы (карбонила) >C=O и гидроксильной группы (гидроксила) -OH, взаимно влияющих друг на друга.
В карбоксильной группе все связи полярны. Атом углерода находится в состоянии sp2-гибридизации. Он образует три σ-связи: две с атомами кислорода и одну с атомом водорода.
Карбоксилат-ион — это сопряжённое основание карбоновой кислоты, RCOO- (или RCO−2). В карбоксилат-ионе оба атома кислорода равноценны, отрицательный заряд равномерно распределён между ними.
Карбоновые кислоты — это слабые кислоты, которые могут отдавать протон (H+) из карбоксильной группы. Обычно они более кислые, чем спирты, но менее кислые, чем минеральные кислоты (такие как соляная кислота).
Кислотность карбоновых кислот обусловлена стабильностью образующегося карбоксилат-иона, который образуется после потери протона.
Для карбоновых кислот значения рКа лежат в интервале 4,2–4,9. Эти кислоты обладают существенно более высокой кислотностью, чем фенолы и тиолы.
Производные карбоновых кислот: сложные эфиры и тиоэфиры (S-эфиры карбоновых кислот), галогенангидриды, ангидриды, амиды, нитрилы, их получение и взаимопревращения.
Карбоновые кислоты при нагревании в присутствии кислотного катализатора реагируют со спиртами, образуя сложные эфиры. Эта реакция получила название «реакции этерификации»:
![]()
Реакция обратима, бразовавшийся сложный эфир в кислой среде подвергается гидролизу до исходных кислоты и спирта. Для смещения равновесия в сторону образования сложного эфира либо используют избыток одного из реагентов (обычно спирта), либо удаляют из реакционной среды воду. Легче всего сложные эфиры образуются из первичных спиртов и низших карбоновых кислот. Вторичные спирты и высшие кислоты реагируют медленнее. Третичные спирты, из-за пространственных препятствий, вступают в реакцию этерификации с большим трудом. кроме того, под действием минеральных кислот третичные спирты легко подвергаются внутримолекулярной дегидратации с образованием алкенов. каталитическое действие серной кислоты состоит в активировании молекулы карбоновой кислоты. При действии на карбоновые кислоты фосфора (III), фосфора (V) галогенидов, тионилхлорида или других галогенирующих реагентов образуются галогенангидриды карбоновых кислот:

Для получения хлорангидридов чаще используют тионилхлорид, так как в этом случае образуются газообразные побочные продукты. Галогенангидриды карбоновых кислот — весьма реакционноспособные вещества, широко применяемые в органическом синтезе. Карбоновые кислоты при нагревании в присутствии водоотнимающих средств фосфора (V) оксида Р2О5 или трифторуксусного ангидрида (CF3CO)2O подвергаются межмолекулярной дегидратации с образованием ангидридов:

Из-за жестких условий протекания реакции этот метод образования амидов редко используют в препаративных целях.
Кислотный и щелочной гидролиз сложных эфиров и амидов.
Сложные эфиры подвергаются гидролизу в кислой и щелочной среде. Кислотный гидролиз представляет собой последовательность обратимых превращений, противоположных реакции этерификации.
Механизм кислотного гидролиза включает протонирование атома кислорода карбонильной группы с последующим образованием карбокатиона, который затем реагирует с молекулой воды. В присутствии водных растворов щелочей сложные эфиры гидролизуются с образованием соли карбоновой кислоты и спирта или фенола.
В отличие от кислотного гидролиза щелочной гидролиз сложных эфиров представляет собой необратимый процесс. его механизм заключается в нуклеофильной атаке гидроксид-ионом атома углерода карбонильной группы с образованием промежуточного аниона, который, отщепляя алкоксид-ион, превращается в молекулу карбоновой кислоты. на заключительной стадии реакции алкоксид-ион, обладая сильными основными свойствами, отрывает протон от образовавшейся молекулы кислоты и превращается в молекулу спирта. При щелочном гидролизе щелочь выступает не в роли катализатора, а в роли реагента. Щелочной гидролиз сложных эфиров часто называют омылением. Это название связано с тем, что при щелочном гидролизе жиров, представляющих собой сложные эфиры глицерина и высокомолекулярных карбоновых кислот, образуются мыла. Амиды гидролизуются намного труднее, чем другие функциональные производные карбоновых кислот. в нейтральной среде гидролиз идет очень медленно. в присутствии минеральных кислот или щелочей амиды гидролизуются довольно легко. в щелочной среде амиды превращаются в соль карбоновой кислоты и аммиак или амины.
В процессе гидролиза в кислой среде амиды образуют карбоновые кислоты и аммониевые соли. Под действием минеральной кислоты сначала образуется протонированная форма амида, которая затем вступает в реакцию с водой.
Реакции ацилирования, этерификации, аминирования и восстановления карбоновых кислот и их производных.
Карбоновые кислоты при нагревании в присутствии кислотного катализатора реагируют со спиртами, образуя сложные эфиры. Эта реакция получила название «реакции этерификации»: Реакция обратима, бразовавшийся сложный эфир в кислой среде подвергается гидролизу до исходных кислоты и спирта. Для смещения равновесия в сторону образования сложного эфира либо используют избыток одного из реагентов (обычно спирта), либо удаляют из реакционной среды воду. Легче всего сложные эфиры образуются из первичных спиртов и низших карбоновых кислот. Вторичные спирты и высшие кислоты реагируют медленнее. Третичные спирты, из-за пространственных препятствий, вступают в реакцию этерификации с большим трудом. кроме того, под действием минеральных кислот третичные спирты легко подвергаются внутримолекулярной дегидратации с образованием алкенов. каталитическое действие серной кислоты состоит в активировании молекулы карбоновой кислоты. Монокарбоновые кислоты, в зависимости от условий, восстанавливаются до альдегидов или первичных спиртов. Поскольку в процессе реакций нуклеофильного замещения в молекулу нуклеофильного реагента вводится ацильная группа, галогенангидриды являются ацилирующими реагентами, а реакции называют реакциями ацилирования. Со слабыми нуклеофильными реагентами, такими, как арены, галогенангидриды реагируют в присутствии кислот льюиса (AlCl3, FeBr3, SnCl2 и др.). Кислоты Льюиса активируют молекулу ацилгалогенида за счет образования донорноакцепторного комплекса (n-комплекса) или иона ацилия. Ацилгалогениды, содержащие атомы водорода при альфа-углеродном атоме, в присутствии сильных оснований (третичных аминов) отщепляют молекулу галогеноводорода, образуя кетены.
При обработке линейных ангидридов спиртами образуются сложные эфиры.
Циклические ангидриды реагируют со спиртами с образованием неполных эфиров дикарбоновых кислот: При взаимодействии линейных ангидридов с аммиаком, первичными или вторичными аминами образуются амиды карбоновых кислот.
При взаимодействии со спиртами в молекуле сложного эфира происходит замена одного спиртового остатка на другой. Эта реакция получила название «реакции переэтерификации». Переэтерификация катализируется минеральными кислотами или щелочами и является обратимой реакцией. для смещения равновесия вправо отгоняют более летучий спирт. Поэтому практическое значение переэтерификация имеет в том случае, когда в состав исходного сложного эфира входит остаток низкокипящего спирта (часто — метилового). Тогда образующийся в результате переэтерификации метиловый спирт может быть отогнан из реакционной среды. При этом равновесие смещается в сторону конечных продуктов.
Под действием лития алюмогидрида LiAlH4 амиды карбоновых кислот восстанавливаются до аминов. N-замещенные амиды восстанавливают до вторичных или третичных аминов.
При восстановлении нитрилов лития алюмогидридом LiAlH4 или водородом в присутствии катализатора образуются первичные амины:
Жирные кислоты, важнейшие представители (пальмитиновая, стеариновая, олеиновая, линолевая, линоленовая). Жиры, сложные липиды (фосфатидовая кислота и ее производные), мыла.
Жирные кислоты – алифатические карбоновые кислоты. Все жирные кислоты, входящие в состав жиров, делят на две группы: насыщенные и ненасыщенные. Ненасыщенные жирные кислоты, имеющие две и более двойных связей, называют полиненасыщенными. Высшие жирные кислоты практически нерастворимы в воде, но их натриевые или калиевые соли, называемые мылами, образуют в воде мицеллы, стабилизируемые за счет гидрофобных взаимодействий. Мыла обладают свойствами ПАВ. Пальмитиновая кислота (C16H32O2 или C15H31COOH) — cлабая химическая органическая кислота, относящаяся к классу высших предельных карбоновых кислот. При стандартных условиях, пальмитиновая кислота — это бесцветные кристаллическое вещество. Наиболее распространённая в природе жирная кислота. Пальмитиновая кислота входит в состав глицеридов большинства животных жиров и растительных масел. Стеариновая кислота (октадекановая кислота) — одноосновная карбоновая кислота алифатического ряда, соответствующая формуле С17Н35COOH. Белые кристаллы, нерастворимые в воде и растворимые в диэтиловом эфире. Стеариновая кислота — одна из наиболее распространённых в природе жирных кислот, входящая в виде глицеридов в состав липидов, прежде всего триглицеридов жиров животного происхождения, последние выполняют функцию энергетического депо. Олеиновая кислота (цис-9-октадеценовая кислота) СН3(СН2)7СН=СН(СН2)7СООН — мононенасыщенная жирная кислота. Маслянистая жидкость, легче воды, без запаха, без цвета, нерастворима в воде, но растворяется в органических растворителях. Содержится во многих животных жирах в виде сложных эфиров — глицеридов. Линолевая кислота — одноосновная карбоновая кислота с двумя изолированными двойными связями CH3(CH2)3-(CH2CH=CH)2(CH2)7COOH. Относится к омега-6-ненасыщенным жирным кислотам. Светло-жёлтая маслянистая жидкость, нерастворимая в воде, но хорошо растворимая во многих органических растворителях. Линолевая кислота относится к так называемым незаменимым жирным кислотам, необходимым для нормальной жизнедеятельности; в организм человека и животных эти кислоты поступают с пищей, главным образом в виде сложных липидов — триглицеридов и фосфатидов. Линоленовая кислота — одноосновная карбоновая кислота с тремя изолированными двойными связями, является ненасыщенной жирной кислотой группы ω3-ненасыщенных жирных кислот. При комнатной температуре — Бесцветная маслянистая жидкость. Относится к незаменимым жирным кислотам, которые должны поступать с пищей для нормальной жизнедеятельности организма человека, и относится к классу омега-3-ненасыщенных жирных кислот. В виде триглицерида она содержится во многих растительных маслах, например, в перилловом, льняном, облепиховом, горчичном, конопляном, соевом и др. Жиры являются сложными эфирами глицерина и высших алифатических кислот, то есть триацилглицеринами, или триглицеридами. Природные жиры представляют собой главным образом смешанные триацилглицерины. По консистенции жиры могут быть твердыми и жидкими. Твердые жиры содержат преимущественно остатки насыщенных высших жирных кислот. В состав жидких жиров, обычно называемых маслами, входят в основном остатки ненасыщенных кислот. Жиры животного происхождения, как правило, — твердые вещества, растительные жиры — жидкие, исключения составляют рыбий жир, являющийся жидкостью, и масло какао — твердое вещество (при обычных условиях). В состав жиров человеческого организма наиболее часто входят остатки насыщенных (стеариновая, пальмитиновая) и ненасыщенных (арахидоновая, олеиновая, линолевая и линоленовая) высших жирных кислот. насыщенные жирные кислоты поступают в организм с пищей, а также образуются путем биосинтеза. олеиновая, линолевая, линоленовая и арахидоновая кислоты не образуются в организме человека; они поступают только с пищей, поэтому их называют незаменимыми. К сложным липидам относят фосфолипиды и гликолипиды. Фосфолипидами называют фосфорилированные липиды. В результате гидролиза фосфолипидов образуются высшие жирные кислоты (ВЖВ), спирт — глицерин или сфингозин, фосфорная кислота Н3РО4, часто — азотистые основания (аминоспирты — коламин, холин, аминокислота — серин) и др. Фосфолипиды в зависимости от природы спирта, входящего в состав молекулы, подразделяют на две группы: глицерофосфолипиды и сфингомиелины. Глицерофосфолипиды являются производными фосфатидной кислоты. Одна из гидроксильных групп фосфатидных кислот, как правило, этерифицирована азотсодержащим соединением: аминоспиртом (коламином, холином), аминокислотой (серином) или циклическим шестиатомным спиртом инозитолом и др. Общая формула глицерофосфолипидов:

, где R1, R2 — остатки ВЖК; R3 — остаток азотсодержащего соединения (коламина, холина, серина) или инозитола. В ряду глицерофосфолипидов выделяют фосфатидилэтаноламины (кефалины), фосфатидилхолины (лецитины), фосфатидилсерины, фосфатидилинозитолы. Кефалины и лецитины метаболически связаны друг с другом и являются главными липидными компонентами клеток. Дипальмитиллецитин снижает поверхностное натяжение и тем самым препятствует слипанию внутренних поверхностей дыхательных путей в легких. Его отсутствие в легких недоношенных новорожденных приводит к развитию синдрома дыхательной недостаточности. Глицерофосфолипиды являются основным компонентом клеточных мембран. Клеточная мембрана состоит преимущественно из глицерофосфолипидов, построенных в виде липидного бислоя толщиной 5,0 нм. Глицерофосфолипиды, подобно мылам, имеют длинную неполярную углеводородную цепь («хвост») и полярную ионную фосфатидную группу («голова»). Неполярные группы ориентированы к центру бислоя, а полярные — по внешним сторонам. Сфингомиелины — другая группа фосфолипидов. в результате гидролиза сфингомиелинов образуются двухатомный ненасыщенный аминоспирт — сфингозин, высшая жирная кислота, фосфорная кислота и холин. N-ацилированный высшими жирными кислотами сфингозин называют церамид. Сфингомиелины содержатся в мембранах животных и растительных клеток, нервной ткани, в ткани печени, почек и других органов. Соли высших жирных кислот называют мылами, а реакцию щелочного гидролиза жиров, при которой образуются мыла,— омылением
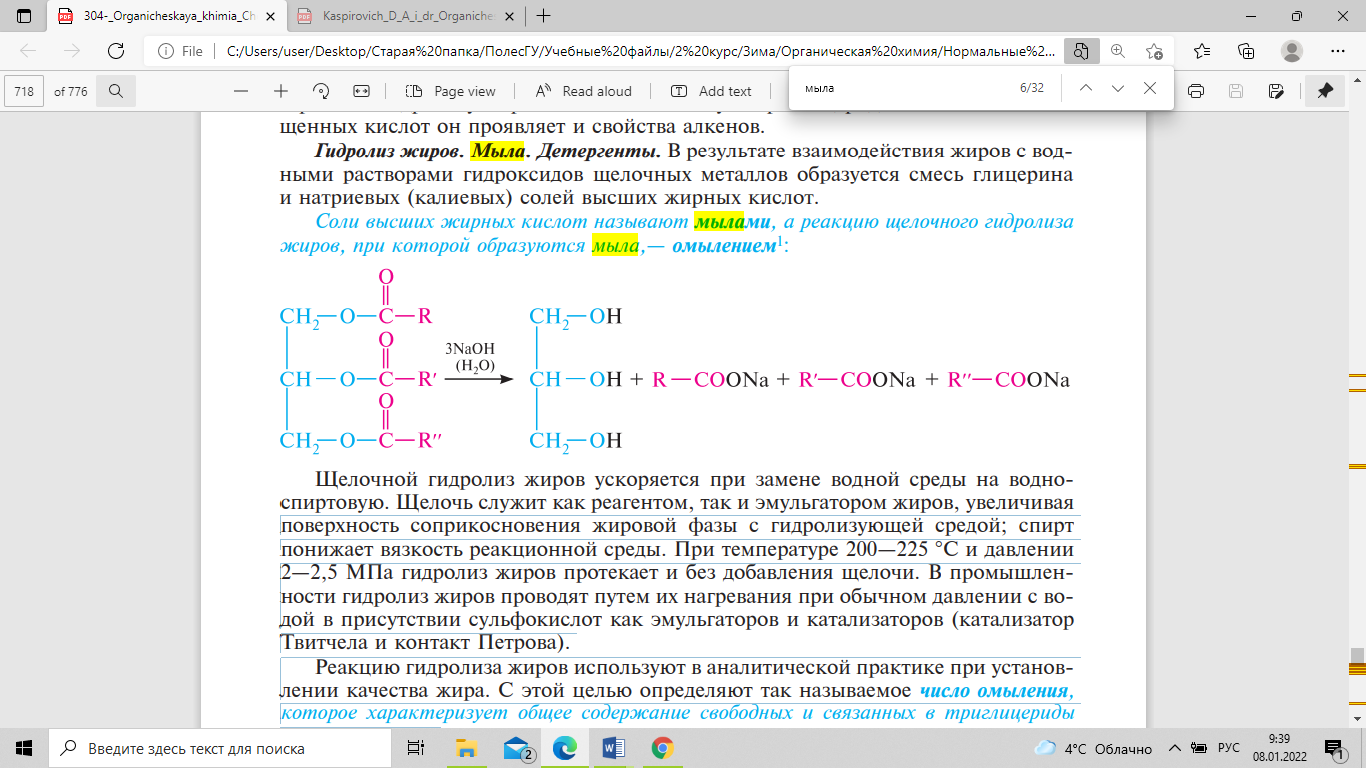
Щелочной гидролиз жиров ускоряется при замене водной среды на водно-спиртовую. Щелочь служит как реагентом, так и эмульгатором жиров, увеличивая поверхность соприкосновения жировой фазы с гидролизующей средой; спирт понижает вязкость реакционной среды. В промышленности гидролиз жиров проводят путем их нагревания при обычном давлении с водой в присутствии сульфокислот как эмульгаторов и катализаторов (катализатор Твитчела и контакт Петрова). Для получения твердого мыла выделившуюся смесь высших жирных кислот нейтрализуют содой. Если нейтрализацию смеси высших жирных кислот проводят с помощью поташа (калия карбоната), то образуется калиевое («зеленое») мыло, отличающееся от натриевого жидкой консистенцией. При нейтрализации жирнокислотной смеси оксидами щелочноземельных и переходных металлов (CaO, MgO, ZnO, PbO и др.) образуются нерастворимые в воде, так называемые «металлические» мыла, которые применяются в качестве медицинских пластырей (например, простой свинцовый). По наличию в молекулах карбоксилатных групп мыла относят к анионным ПАВ. Описанные свойства мыл обусловливают их моющее действие. Вода, содержащая мыло, благодаря уменьшению своего поверхностного натяжения, приобретает способность проникать в тончайшие поры на отмываемой поверхности. При наличии на этой поверхности частиц веществ, не смачивающихся водой (жиров, восков, нефтепродуктов и пр.), молекулы мыла сцепляются с гидрофобными частицами загрязнений своими углеводородными «хвостами», образуя вокруг таких частиц плотную изолирующую пленку, вклиниваются между загрязняющей частицей и очищаемой поверхностью и в конечном итоге отрывают эту частицу, переводя ее во взвешенное состояние в толще водной среды.
Ненасыщенные карбоновые кислоты: акриловая, метакриловая, полимеры на их основе.
Ненасыщенными монокарбоновыми кислотами называют монокарбоновые кислоты, содержащие в углеводородном радикале кратную связь. Акриловая кислота Н2С=СН—СООН. Бесцветная жидкость с резким запахом (т. пл. 13 °с, т. кип. 141 °с), хорошо растворяется в воде, легко полимеризуется с образованием полиакриловой кислоты. Важное практическое значение имеют полимеры на основе сложных эфиров акриловой кислоты (полиакрилаты):

В медицине полиакрилаты находят применение при изготовлении зубных протезов. Метакриловая кислота (2-метилпропеновая кислота). Бесцветная жидкость (т. кип. 160,5 °с). Метакриловая кислота легко полимеризуется. важное значение имеет метиловый эфир метакриловой кислоты, полимеризацией которого получают полиметилметакрилат — органическое стекло (плексиглас): Области применения: осветительная техника (плафоны, перегородки, лицевые экраны, рассеиватели), наружная реклама (лицевые стёкла для коробов, световых букв, формованные объёмные изделия), торговое оборудование (подставки, витрины, ценники), сантехника (оборудование ванных комнат), строительство и архитектура (остекление проёмов, перегородки, купола, танц-пол, объёмные формованные изделия, аквариумы), транспорт (остекление самолётов, катеров, обтекатели), приборостроение (циферблаты, смотровые окна, корпуса, диэлектрические детали, ёмкости).
Дикарбоновые кислоты. Основные представители: щавелевая, малоновая, адипиновая, фталевые кислоты. Фумаровая и малеиновая кислоты. Полиэфирные волокна на основе терефталевой и адипиновой кислот.
Дикарбоновыми кислотами называют производные углеводородов, содержащие в своем составе две карбоксильные группы. Щавелевая кислота НООС—СООН, белое кристаллическое вещество (т. пл. 189 °с), легко растворяется в воде и спиртах, содержится в виде солей во многих растениях (щавель, ревень и др.). Соли и сложные эфиры щавелевой кислоты называют оксалатами. кристаллы кальция оксалата трудно растворимы в воде и могут откладываться при патологических состояниях в почках в виде камней (почечно-каменная болезнь). Впромышленности щавелевую кислоту получают из натрия формиата. Специфическими реакциями щавелевой кислоты являются разложение концентрированной серной кислотой и окисление. При нагревании с концентрированной серной кислотой щавелевая кислота разлагается с образованием углерода (II) оксида, углерода (IV) оксида и воды. При окислении щавелевая кислота образует углерода (IV) оксид и воду. Щавелевую кислоту используют для очистки металлов от ржавчины и накипи, а в качестве протравы — в кожевенном производстве. Применяют также в синтезе бриллиантового зеленого. Малоновая кислота НООС—СН2—СООН, белое кристаллическое вещество (т. пл. 135 °с), растворимое в воде, этаноле, эфире, содержится в соке сахарной свеклы, соли и сложные эфиры малоновой кислоты называют малонатами. В результате электроноакцепторного влияния карбоксильных групп в молекуле малоновой кислоты атомы водорода метиленовой группы приобретают подвижность (сн-кислотность). Поэтому в присутствии оснований малоновая кислота способна образовывать не только дианион с участием двух карбоксильных групп, но и частично — трианион за счет ионизации активной метиленовой группы. с участием метиленовой группы малоновая кислота в присутствии оснований вступает в реакцию конденсации с альдегидами и кетонами, не содержащими атома водорода при атоме углерода в альфа-положении. В результате реакции образуются ненасыщенные монокарбоновые кислоты. Малоновый эфир используется в органическом синтезе для получения моно- и дикарбоновых кислот, при действии на малоновый эфир натрия алкоголята атом водорода метиленовой группы замещается и образуется натриймалоновый эфир, в котором анион весьма устойчив вследствие делокализации заряда за счет карбоксильных групп. Натриймалоновый эфир легко алкилируется, ацилируется и реагирует с другими электрофильными реагентами. При алкилировании образуется алкилмалоновый эфир, который в процессе гидролиза превращается в алкилмалоновую кислоту, алкилмалоновые кислоты легко декарбоксилируются с образованием карбоновых кислот. Адипиновая кислота HOOC(CH2)4COOH, белое кристаллическое вещество (т. пл. 152 °с), малорастворимое в воде. соли и сложные эфиры адипиновой кислоты называют адипинатами. Применяется главным образом в производстве синтетического волокна — найлона, входит в состав противоглистного средства — пиперазина адипината. Фталевая кислота С8H6O4 — cлабая химическая органическая кислота, относящаяся к классу карбоновых кислот ароматического ряда. При стандартных условиях — бесцветные моноклинные кристаллы, почти не имеющие запаха. Токсична. Фталевая кислота декарбоксилируется в присутствии катализаторов при 200 °C до бензойной кислоты, а при 350 °C в присутствии оксида кадмия CdO — до бензола. Как и другие карбоновые кислоты, фталевая кислота образует соли и сложные эфиры по одной или двум карбоксильным группам, называемые фталатами. В больших количествах вещество раздражает слизистые оболочки глаз и кожи, вызывает общетоксическое действие, используются в основном в производстве лекарственных средств, красителей и пластификаторов для поливинилхлорида. Фталаты, имитирующие структуру эстрогена, могут вызывать нарушения в эндокринной системе. Исследования, проводившиеся на животных и людях, доказывают, что фталаты способны ослаблять действие тестостерона — основного мужского гормона позвоночных и человека, стимулирующего функцию мужских половых органов и развитие вторичных половых признаков. Фумаровая кислота HO2CCH=CHCO2H, является транс-изомером, цис-изомер — малеиновая кислота. Кристаллы имеют фруктовый вкус. Соли и эфиры называют фумаратами. Фумаровая кислота в качестве пищевой добавки обозначается E297. Фумарат является интермедиатом в цикле Кребса. Кожа человека образует фумарат при действии солнечного света. Фумарат также является побочным продуктом цикла мочевины. Эфиры фумаровой кислоты применяют для лечения псориаза. Малеиновая кислота HOOC-CH=CH-COOH. Название по номенклатуре IUPAC — цис-бутендиовая кислота. Её транс-изомер называется фумаровой кислотой. Соли и сложные эфиры малеиновой кислоты называются малеаты. Наиболее важным применением малеиновой кислоты является использование её в производстве фумаровой кислоты. Для производства фумаровой кислоты малеиновою кислоту каталитически (катализаторы — тиомочевина, неорганические кислоты) изомеризуют. Очень малая растворимость фумаровой кислоты сильно упрощает её отделение от непрореагировавшей малеиновой кислоты. Эти кислоты являются геометрическими изомерами, они не могут самопроизвольно превращаться одна в другую. Это объясняется тем, что для их взаимопревращение нужно разорвать π связь между атомами углерода. Путём озонолиза малеиновою кислоту превращают в глиоксиловую кислоту. Полиэтилентерефталат (ПЭТФ, лавсан) — термопластик, наиболее распространённый представитель класса полиэфиров. Продукт поликонденсации этиленгликоля с терефталевой кислотой (или её диметиловым эфиром); твёрдое, бесцветное, прозрачное вещество в аморфном состоянии и белое, непрозрачное в кристаллическом состоянии. Переходит в прозрачное состояние при нагреве до температуры стеклования и остаётся в нём при резком охлаждении и быстром проходе через т. н. «зону кристаллизации». Одним из важных параметров ПЭТ является характеристическая вязкость, определяемая длиной молекулы полимера. С увеличением присущей вязкости скорость кристаллизации снижается. Прочен, износостоек, хороший диэлектрик. Распространению бутылок из ПЭТ способствовала их сравнительная дешевизна и практичность. Самое массовое из всех видов химических волокон для бытовых целей (одежда) и техники; металлизированная плёнка широко используется в качестве декоративного, термоизоляционного, светоотражающего, архитектурно-строительного материала. Нейлон — семейство синтетических полиамидов, используемых преимущественно в производстве волокон. Формула волокна из анида: [—HN(CH2)6NHOC(CH2)4CO—]n[1]. Анид синтезируется поликонденсацией адипиновой кислоты и гексаметилендиамина. Пластмассовые изделия могут изготавливаться из жёсткого нейлона — эколона, путём впрыскивания в форму жидкого нейлона под большим давлением, чем достигается бо́льшая плотность материала. В промышленности нейлон применяется для изготовления втулок, вкладышей, плёнок и тонких покрытий. Нейлон, нанесённый на трущиеся поверхности в виде облицовки или тонкослойного покрытия на тонкие металлические втулки, вкладыши и корпуса подшипников, повышает их эксплуатационные качества. Используется также для изготовления струн некоторых музыкальных инструментов как альтернатива традиционным струнам животного происхождения.
Классификация углеводов. Биологическая Роль и распространенность углеводов.
Термин «углеводы» предложен в 1844 году русским химиком Шмидтом на основании данных элементного состава первых представителей этого класса соединений, так как было установлено, что их молекулы состоят из атомов углерода, водорода и кислорода, причем соотношение атомов водорода и кислорода такое же, как в молекуле воды [Cx(H2O)y]. Углеводы (сахара, карбогидраты) — обширная группа природных и синтетических соединений, являющихся по химическому строению полигидроксильными веществами, содержащими альдегидную или кетонную группы, или образующие их в результате гидролиза. Углеводы (сахара) составляют основную массу органических веществ нашей планеты. В растительном и животном мире они широко распространены главным образом в виде различных производных и значительно реже — в свободном виде. Углеводы, выполняя различные жизненно важные функции, служат исходным материалом для биосинтеза многих органических соединений живых организмов. В природе углеводы образуются в результате фотосинтеза, осуществляемого растениями с участием углерода (IV) оксида, воды и поглощающих солнечный свет пигментов (хлорофилл и др.). Для растений одни виды углеводов служат строительным материалом, выполняя опорную функцию (целлюлоза), другие — источником резервной энергии (крахмал, инулин). для человека и животных углеводы являются продуктами питания с высокой энергетической ценностью. в организме крахмал, дисахариды, а в некоторых случаях и целлюлоза под влиянием ферментов распадаются с образованием в основном глюкозы, которая окисляется в тканях до углерода (IV) оксида и воды с выделением энергии. избыток глюкозы превращается в гликоген, запасаемый в печени и мышцах. Гликоген снабжает организм глюкозой при выполнении физических нагрузок, а также при недостатке или отсутствии пищи. Углеводы входят в состав гликолипидов, гликопротеидов, нуклеотидов, нуклеозидов и нуклеиновых кислот, составляющих, как известно, основу живой материи. Углеводы составляют сырьевую базу для текстильной, целлюлозно-бумажной, пищевой, деревообрабатывающей и других отраслей промышленности. Углеводы делят на простые и сложные в зависимости от количества моносахаридных единиц, связанных в молекулу. Простые углеводы, или моносахариды (монозы), не способны гидролизоваться. Сложные углеводы при гидролизе образуют моносахариды. Сложные углеводы, в свою очередь, хотя и условно, подразделяют на олигосахариды, образующие при гидролизе от двух до десяти молекул моносахаридов, и полисахариды (полиозы), гидролизующиеся с образованием более десяти молекул моносахаридов.
Моносахариды (рибоза, дезоксирибоза, глюкоза, фруктоза). Стереохимия моносахаридов, D- и L-ряды. Кольчато-цепная таутомерия. Пиранозные и фуранозные формы. a- и bb-аномеры. Мутаротация. Эпимеризация.
Моносахариды (монозы) — полигидроксильные соединения, содержащие альдегидную или кетонную группы. Моносахариды, за исключением глюкозы и фруктозы, редко встречаются в природе в свободном виде, в основном они входят в состав олиго- и полисахаридов, гликозидов, гликолипидов, нуклеозидов и других высокомолекулярных соединений. Молекулы моносахаридов содержат несколько асимметрических атомов углерода и поэтому существуют в виде различных пространственных изомеров. Альдопентоза имеет три асимметрических атома углерода, а следовательно, одной и той же структурной формуле соответствует восемь стереоизомеров (23), альдогексоза содержит четыре асимметрических углеродных атома и может существовать в виде 16 стереоизомеров (24) (N = 2n, где n — количество асимметрических атомов углерода). Изомеры моносахаридов подразделяют на D- и L-стереохимические ряды, принадлежность к которым определяется по конфигурации асимметрического атома углерода, максимально удаленного от карбонильной группы (для пентоз — с-4, для гексоз — с-5). Если конфигурация этого хирального атома углерода совпадает с конфигурацией D-глицеринового альдегида, то моносахарид относят к D-ряду, если же с конфигурацией L-глицеринового альдегида — то к L-ряду:

Представители D-ряда являются оптическими антиподами L-ряда, то есть альдогексоза существует в виде восьми пар энантиомеров. D-Глюкоза и L-глюкоза являются энантиомерами, большинство природных моносахаридов относится к D-ряду. Пространственные изомеры моносахаридов, отличающиеся конфигурацией одного или нескольких атомов углерода и не являющиеся зеркальными изомерами (энантиомерами), называются диастереомерами. так, D-глюкоза и D-галактоза, D-манноза и D-глюкоза, D-манноза и D-галактоза составляют пары диастереомеров. Моносахариды обладают оптической активностью. Вращение плоскости поляризованного света вправо обозначают знаком (+), а влево — знаком (–). Однако следует помнить, что направление вращения определяется экспериментально и никаким образом не связано с конфигурацией, то есть принадлежностью к D- или L-ряду. так, встречающаяся в природе форма D-глюкозы является правовращающей, а природная форма D-фруктозы — левовращающей. Моносахариды, являясь полигидроксиальдегидами или полигидроксикетонами, образуют циклические полуацетали в результате внутримолекулярного взаимодействия карбонильной и пространственно сближенной с ней спиртовой групп. Причем, в соответствии с теорией напряжения циклов, наиболее благоприятно взаимодействие, если оно приводит к образованию пяти- или шестичленных циклов. При взаимодействии оксогруппы с гидроксильной группой при с-5 альдогексоз или с-6 кетогексоз образуется пиранозный шестичленный цикл (от названия шестичленного гетероцикла пиран + окончание -оза). При взаимодействии оксогруппы с гидроксильной группой при с-4 альдогексоз или с-5 кетогексоз образуется фуранозный пятичленный цикл (от названия пятичленного гетероцикла фуран + окончание -оза). Аномеры — диастереомеры, отличающиеся конфигурацией аномерного атома углерода. У альфа-аномера расположение полуацетального гидроксила такое же, как гидроксила «концевого» хирального центра (асимметрического атома углерода, определяющего принадлежность к D- или L-ряду). Таким образом, в проекционных формулах моносахаридов D-ряда гликозидный гидроксил у альфа-аномера расположен справа от вертикальной линии углеродной цепи, а у бета-аномера — слева. Моносахариды являются таутомерными веществами, в кристаллическом состоянии они имеют циклическое строение. так, D-глюкоза, полученная дробной кристаллизацией из этилового спирта или воды, находится в форме альфа-D-глюкопиранозы. в водном растворе циклическая форма под влиянием растворителя превращается через открытую оксоформу в другие циклические формы — пиранозные и фуранозные с альфа- и бета-конфигурацией аномерного центра. Таким образом, в водном растворе моносахариды существуют в виде пяти таутомерных форм — открытой, альфа- и бета-пиранозных и альфа- и бета-фуранозных. Такой вид таутомерии называется кольчато-цепной, или цикло-оксо-таутомерией. Cпособность моносахаридов к цикло-оксо-таутомерии объясняет выявленное задолго до установления их строения явление мутаротации — самопроизвольного изменения величины оптического вращения свежеприготовленных растворов оптически активных соединений. В свежеприготовленном водном растворе глюкозы наблюдается уменьшение угла оптического вращения с +112,2° до установления постоянного значения +52,5°, химической основой этого процесса является цикло-оксо-таутомерия. В разбавленных растворах щелочей при комнатной температуре моносахариды подвергаются изомеризации с образованием равновесной смеси моноз, различающихся конфигурацией углеродных атомов C-1 и с-2. так, D-глюкоза, выдержанная в растворе натрия гидроксида (8•10–3) при 35 °с в течение 4 суток, превращается в смесь, состоящую из D-фруктозы (~ 28 %), D-маннозы (~ 3 %) и D-глюкозы (~ 69 %). аналогичная изомеризация наблюдается у кетоз (фруктозы). Изомерные превращения моносахаридов под действием щелочей называют эпимеризацией и приводят к образованию эпимеров - диастереоизомеров, отличающиеся конфигурацией только одного из нескольких хиральных центров (например, D-глюкоза и D-манноза, D-ксилоза и D-рибоза и др.). Взаимопревращения в слабощелочной среде протекает через ендиольную форму, которая образуется в результате миграции протона от альфа-углеродного атома к карбонильной группе. При обратном превращении ендиольной формы в карбонильную образуется смесь трех моносахаридов. В бета-фуранозной форме D-рибоза входит в состав РНК, ряда коферментов, гликозидов и антибиотиков. Дезоксирибоза C5H10O4 — углевод, альдопентоза: моносахарид, содержащий пять атомов углерода и альдегидную группу в линейной структуре. Это дезоксисахар — производное рибозы, где гидроксильная группа у второго атома углерода замещена водородом с потерей атома кислорода (дезокси — отсутствие атома кислорода). Рибоза формирует пятичленное кольцо, состоящее из четырёх атомов углерода и атома кислорода. Гидроксильные группы соединены с тремя атомами углерода. Последний атом углерода и гидроксильная группа связаны с одним из атомов углерода, соединённых с кислородом. В дезоксирибозе атом углерода, расположенный дальше всего от атома кислорода, лишён гидроксильной группы. Входит в состав ДНК, вместе с азотистым основанием и остатком фосфорной кислоты образуя мономерную единицу дезоксирибонуклеиновой кислоты — нуклеотид. D-Глюкоза (виноградный сахар, декстроза), широко распространена в природе: в свободном состоянии содержится в растениях, меде, крови; входит в состав многих дисахаридов (лактоза, сахароза и др.); полисахаридов (крахмал, клетчатка, гликоген и др.). Глюкоза — главный источник энергии для большинства организмов. Получают гидролизом крахмала или целлюлозы в присутствии минеральных кислот. Глюкоза используется в качестве сырья для производства витамина с и лекарственного препарата «кальция глюконата»; в медицине применяется в виде растворов для внутривенного введения при гипогликемии, инфекционных заболеваниях, заболеваниях печени и так далее; является компонентом различных кровезаменителей и противошоковых жидкостей. Под действием ферментов глюкоза подвергается брожению. известно много видов брожения — спиртовое, молочнокислое, маслянокислое, лимоннокислое и др. наиболее важным из них является спиртовое брожение, которое происходит под влиянием фермента дрожжей — зимазы. Этот вид брожения используют в промышленности для получения этанола, а также в виноделии и пивоварении. D-Фруктоза (плодовый или фруктовый сахар, левулеза), в свободном состоянии содержится во фруктах, меде; входит в состав ряда олигосахаридов (сахароза, раффиноза) и полисахаридов (инулин). Фосфаты D-фруктозы — промежуточные продукты энергетического обмена углеводов в живых организмах. фруктоза слаще глюкозы и сахарозы. Кристаллическая фруктоза представляет собой фруктопиранозу. В состав олиго- и полисахаридов обычно входит в фуранозной форме. Получают фруктозу гидролизом инулина, содержащегося в клубнях георгинов, корнях цикория.
Гликозиды. Особые свойства гликозидного гидроксила. Реакции окисления и восстановления глюкозы. Глюконовая, глюкаровая и глюкуроновая кислоты. Глюцит (сорбит), ксилит. Реакции алкилирования и ацилирования моносахаридов. Аскорбиновая кислота (витамин С).
Гликозиды — органические соединения, молекулы которых состоят из двух частей: углеводного (пиранозидного или фуранозидного) остатка и неуглеводного фрагмента (агликона). В качестве гликозидов в более общем смысле могут рассматриваться и углеводы, состоящие из двух или более моносахаридных остатков.
Чаще всего гликозиды встречаются в листьях и цветках растений, реже в других органах. В состав гликозидов входят углерод, водород, кислород, реже азот и только некоторые содержат серу.
Многие из гликозидов токсичны или обладают сильным физиологическим действием. Например, гликозиды наперстянки, строфанта и другие.
Гликозидный гидроксил отличается от других гидроксильных групп моносахарида повышенной реакционной способностью, так как находится у атома углерода, имеющего две связи с атомами кислорода. Это приводит к поляризации связи О–Н и увеличению подвижности атома водорода.
Особые свойства гликозидного гидроксила:
легко замещается на другие нуклеофилы, в результате чего образуются различные производные углеводов по С–1: простые и сложные эфиры, галогениды и др.;
углеродный атом при гликозидном гидроксиле легко меняет конфигурацию (явление мутаротации).
Суммарное уравнение реакции окисления глюкозы выглядит так: C 6 H 12 O 6 + 6O 2 + 38АДФ + 38H 3 PO 4 = 6CO 2 + 44H 2 O + 38АТФ. Распад глюкозы по аэробному пути (аэробный гликолиз) дает энергию для восстановления 38 молекул АТФ.
Реакция «серебряного зеркала» C 5 H 11 O 5– CH=O + 2Ag 2 O C 5 H 11 O 5 COOH + 2Ag↓ Восстановление. Восстановление глюкозы СН 2 ОН– (СНОН) 4– СН=О + Н 2 СН 2. ОН– (СНОН) 4– СН 2– ОН (сорбит). Сорбит используется как заменитель сахара и глюкозы, поскольку он хуже усваивается организмом и не так вреден как избыток сахара. Его добавляют в жевательную резинку и в диетические продукты.

Глюконовая кислота — монокарбоновая кислота, которая образуется при окислении глюкозы бромной водой. В медицине используется её кальциевая соль — глюконат кальция.
Глюкаровая (или сахарная) кислота — дикарбоновая кислота, которая получается при окислении глюкозы азотной кислотой.
Глюкуроновая кислота — сахарная кислота, полученная из глюкозы, шестой атом углерода которой окислен до карбоновой кислоты. Глюкуроновая кислота вырабатывается в печени человека и большинства животных. Она может связываться с такими веществами, как гормоны, лекарства и токсины, чтобы облегчить их перенос по телу.
Сорбит (сорбитол) — многоатомный спирт натурального происхождения. Содержится в плодах рябины, яблоках, абрикосах. Коэффициент сладости — 0,6, в 4 раза менее калориен, нежели сахар. Иногда сорбит добавляют в соки и прохладительные напитки в качестве консерванта.
Ксилит — получают из отходов переработки кукурузы и хлопковых семян. Коэффициент сладости — 1,0. Энергетическая ценность и сладость аналогичны сахару, однако ксилит не оказывает разрушительного влияния на состояние эмали зубов, предотвращает развитие кариеса, потому входит в состав некоторых зубных паст и жевательных резинок. Повышает секрецию желудочного сока, обладает желчегонным и слабительным действиями. Максимально допустимая суточная доза — 40–50 г в сутки.
Перед употреблением сахарозаменителей рекомендуется проконсультироваться с врачом.
Алкилирование моносахаридов — действие алкилирующих агентов на моносахариды приводит к образованию неполных и полных простых эфиров. При этом наиболее активно взаимодействует полуацетальный гидроксил. В результате образуется простой эфир — гликозид (в случае глюкозы — глюкозид). Связь алкила с углеродом (через кислородный мостик) называют гликозидной связью.
Ацилирование моносахаридов — при действии на моносахариды или сахараты ангидридов кислот или других ацилирующих агентов образуются сложные эфиры циклических форм моносахаридов. Например, при нагревании уксусного ангидрида с глюкозой образуется пента-О-ацетилглюкоза.
Аскорбиновая кислота (витамин С) — водорастворимое органическое соединение, одно из основных веществ, необходимых человеку для поддержания жизнедеятельности. Не синтезируется организмом, а поступает только извне с продуктами питания.
Свойства витамина С:
защищает клетки и ткани внутренних органов от повреждений;
регулирует окислительно-восстановительные процессы;
способствует синтезу коллагена, проколлагена, стероидных и нейрогормонов, медиаторов;
участвует в обмене фолиевой кислоты и железа.
Суточная потребность в витамине С зависит от ряда причин: возраста, пола, беременности, климатических условий, вредных привычек. Средняя суточная доза витамина С — 70–100 мг.
Применение:
профилактика и лечение гипо- и авитаминоза витамина С;
в период восстановления после тяжёлых заболеваний, лихорадочных состояний на фоне острых респираторных заболеваний;
в комплексной терапии при геморрагических диатезах, кровотечениях, вяло заживающих ранах и переломах костей.
Дисахариды И их типы (восстанавливающие И невосстанавливающие). Сахароза, лактоза, мальтоза, целлобиоза.
Дисахаридами называют углеводы, молекулы которых состоят из двух остатков моносахаридов одинаковой или разной природы, соединенных между собой гликозидной связью. Являясь O-гликозидами, дисахариды легко гидролизуются в кислой среде с образованием двух молекул моносахаридов. в зависимости от способа образования гликозидной связи, дисахариды разделяют на две группы — восстанавливающие и невосстанавливающие. В восстанавливающих дисахаридах гликозидная связь образуется за счет полуацетальной (гликозидной) гидроксильной группы одного и любой спиртовой гидроксильной группы (чаще у С-4) другого моносахарида. При этом в молекуле остается одна свободная полуацетальная гидроксильная группа, вследствие чего дисахарид сохраняет способность к цикло-оксо-таутомерии и, следовательно, обладает восстанавливающими свойствами. В свежеприготовленных растворах таких дисахаридов наблюдается явление мутаротации. Представителями восстанавливающих дисахаридов является мальтоза, целлобиоза, лактоза. Мальтоза (солодовый сахар). Молекула мальтозы состоит из двух остатков D-глюкопиранозы, связанных 1,4-гликозидной связью. При этом остаток глюкозы, аномерный атом углерода которого участвует в образовании гликозидной связи, находится в альфа-форме, а остаток глюкозы со свободной полуацетальной гидроксильной группой может иметь α-конфигурацию (α-мальтоза) или β-конфигурацию (β-мальтоза). Растворы мальтозы способны к мутаротации. Мальтоза является восстанавливающим дисахаридом и дает положительные реакции с реактивом Толленса и реактивом Фелинга. С участием альдегидной формы мальтоза вступает в характерные для моносахаридов реакции. При окислении в мягких условиях (под действием бромной воды) мальтоза превращается в мальтобионовую кислоту. С участием циклических форм мальтоза, подобно моносахаридам, подвергается алкилированию и ацилированию. При взаимодействии мальтозы с такими слабыми алкилирующими реагентами, как спирты (в присутствии HCl), образуются гликозиды. Реакция протекает за счет полуацетального гидроксила. При взаимодействии мальтозы с избытком сильных алкилирующих реагентов (алкилгалогенидов, диалкилсульфатов (RO)2SO2 и др.) осуществляется алкилирование по всем гидроксильным группам. Мальтоза подвергается ацилированию также при участии всех гидроксильных групп. Мальтоза содержится в небольших количествах в некоторых растениях, образуется при ферментативном гидролизе крахмала, легко растворяется в воде, водные растворы имеют сладкий вкус. В организме человека расщепляется до D-глюкозы. Целлобиоза. Молекула целлобиозы, как и мальтозы, состоит из двух остатков D-глюкопиранозы, связанных 1,4-гликозидной связью, но, в отличие от мальтозы, в молекуле целлобиозы остаток глюкозы, полуацетальный гидроксил которого участвует в образовании гликозидной связи, имеет β-конфигурацию. Остаток же глюкозы со свободной полуацетальной группой, аналогично мальтозе, может иметь α-конфигурацию (α-целлобиоза) или β-конфигурацию (β-целлобиоза). Растворы целлобиозы способны мутаротировать, целлобиоза является восстанавливающим дисахаридом и дает положительные реакции с реактивами Толленса и Фелинга. С участием альдегидной формы вступает в характерные для моносахаридов реакции. При окислении в мягких условиях образуется целлобионовая кислота, с участием циклических форм подвергается алкилированию и ацилированию. Является бесцветным кристаллическим веществом, легко растворяется в воде. она не расщепляется в организме человека и поэтому не может быть использована в качестве продукта питания. Лактоза (молочный сахар). Молекула состоит из остатков D-галактопиранозы и D-глюкопиранозы, связанных 1,4-гликозидной связью. В образовании гликозидной связи принимает участие полуацетальный гидроксил D-галактопиранозы. Растворы мутаротируют и дают положительную реакцию с реактивом Толленса и реактивом Фелинга. При окислении лактозы в мягких условиях образуется лактобионовая кислота. для лактозы характерен ряд других реакций, присущий восстанавливающим дисахаридам. Содержится в молоке, не подвергается спиртовому брожению, по сравнению с сахарозой обладает меньшей сладкостью (в 4—5 раз). При кислотном или ферментативном гидролизе лактозы образуется D-глюкоза и D-галактоза, обладает низкой гигроскопичностью, применяется в фармации при изготовлении порошков и таблеток. В молекулах невосстанавливающих дисахаридов гликозидная связь образуется за счет полуацетальных гидроксильных групп обоих моносахаридов. такие дисахариды не имеют в своем составе свободного полуацетального гидроксила, поэтому в растворах они существуют только в циклической форме, их растворы не мутаротируют и не обладают восстанавливающими свойствами. невосстанавливающие дисахариды не дают реакций по альдегидной группе и гликозидному гидроксилу, они способны лишь к образованию простых и сложных эфиров. Представителем невосстанавливающих дисахаридов является сахароза. Сахароза (тростниковый или свекловичный сахар). Молекула сахарозы состоит из остатков D-глюкозы и D-фруктозы. Представляет собой бесцветное кристаллическое вещество, хорошо растворяется в воде, имеет сладкий вкус. растворы сахарозы оптически активны. Под действием минеральных кислот при нагревании или ферментов сахароза гидролизуется с образованием смеси D-глюкозы и D-фруктозы. При этом происходит изменение знака удельного вращения, то есть характерное для сахарозы вращение плоскости поляризации вправо. В связи с изменением в процессе гидролиза сахарозы знака удельного вращения гидролиз сахарозы получил название «инверсии». Отсюда образующаяся в процессе гидролиза смесь равных количеств D-глюкозы и D-фруктозы называется инвертным сахаром. инвертный сахар является основной составной частью пчелиного меда. Причиной инверсии сахарозы является относительно большое удельное вращение D-фруктозы влево, чем D-глюкозы вправо, поэтому образующаяся при гидролизе смесь обладает левым вращением. Сахароза содержится в сахарном тростнике и сахарной свекле (17—20 %), из которых ее получают в промышленности. в фармации сахарозу используют для приготовления порошков, сиропов, микстур и др.
Полисахариды (крахмал, целлюлоза, хитин, гликоген).
К полисахаридам относят соединения, молекулы которых содержат более десяти моносахаридных звеньев, связанных о-гликозидной связью. Чаще всего полисахариды состоят из нескольких сотен и даже тысяч моносахаридных остатков, образующих линейные или разветвленные полимерные цепи. Гликозидные связи в полисахаридах, как правило, образуются за счет гликозидного гидроксила одного и спиртового гидроксила другого моносахаридных остатков. В большинстве своем эти связи возникают между С-1 и С-2, С-1 и С-3 или С-1 и С-6. На конце полисахаридной цепи находится восстанавливающий остаток моносахарида, но поскольку его доля в молекуле ничтожна, то полисахариды с большой молекулярной массой практически не обладают восстанавливающей способностью. Если в состав полисахаридов входят остатки только одного моносахарида, то их называют гомополисахаридами. Полисахариды, состоящие из разных моносахаридных единиц, называют гетерополисахаридами. Гомополисахариды, построенные из остатков пентоз, называются пентозанами, а из остатков гексоз — гексозанами. Общая формула пентозанов — (с5н8о4)n, а гексозанов — (с6н10о5)n. Подавляющее большинство природных полисахаридов — гексозаны (крахмал, целлюлоза, гликоген, декстраны и др). Крахмал служит основным источником резервной энергии в растениях; встречается главным образом в семенах, клубнях, корнях. Содержит примерно 20 % растворимой в воде фракции, называемой амилозой, и около 80 % нерастворимой фракции, называемой амилопектином. При постепенном кислотном и ферментативном гидролизе амилоза и амилопектин расщепляются до декстринов (смесь полисахаридов с меньшей молекулярной массой), дальнейший гидролиз которых приводит к мальтозе, а затем к D-глюкозе. Различие в строении амилозы и амилопектина обусловлено характером гликозидных связей. Амилоза — линейный полимер, содержащий более 1000 мономерных звеньев, в котором D-глюкопиранозные остатки связаны альфа-1,4-гликозидной связью. Молекулярная масса амилозы составляет примерно 150 000—600 000, ее молекулы обладают гибкостью и могут принимать различные пространственные формы. В присутствии комплексообразователей, например йода, она может существовать в виде спирали, в каждом витке которой содержится шесть остатков глюкозы. Размер внутренней полости спирали позволяет разместиться в ней молекуле йода, что приводит к образованию окрашенного в синий цвет комплекса. На этом свойстве основано использование крахмала в фармацевтическом анализе в качестве индикатора. Амилопектин — полимер разветвленной структуры, который может содержать приблизительно 600—5000 остатков D-глюкозы в молекуле. Молекулярная масса амилопектина достигает 1—6 млн. Все цепи полисахарида — основная и боковые — построены однотипно: остатки глюкозы в них соединены альфа-1,4-гликозидной связью, боковые ответвления связаны с основной цепью альфа-1,6-гликозидной связью. Между двумя соседними точками ветвления основная цепь содержит 20—25 моносахаридных остатков. В связи с наличием большого числа ответвлений молекула амилопектина не способна принимать конформацию спирали и связывает йод лишь в незначительном количестве с образованием красного окрашивания. Крахмал служит основным источником углеводов в пищевом рационе человека. фермент амилаза, содержащийся в слюне, расщепляет альфа-гликозидные связи крахмала до декстринов и частично — до мальтозы, дальнейший распад которых до глюкозы происходит в кишечнике. в фармации крахмал используется в производстве таблеток, а также для приготовления присыпок и паст. Гликоген (животный крахмал). если у большинства растений резервным полисахаридом является крахмал, то в животных организмах эту функцию выполняет гликоген. Этот полисахарид снабжает организм глюкозой при повышенных физических нагрузках и в промежутках между приемами пищи. Гликоген построен аналогично амилопектину, но представляет собой еще более разветвленную структуру. Связь глюкопиранозных остатков в основной и боковых цепях альфа-1,4, а в местах разветвления — альфа-1,6. Между точками разветвления содержится 10—12, реже — 2—4 моносахаридных остатков. Молекулярная масса гликогена может достигать 100 млн и выше. в отличие от большинства других резервных полисахаридов гликоген хорошо растворим в воде. Сильная разветвленность цепей гликогена способствует атаке его ферментами с разных сторон одновременно. Это обстоятельство приводит к чрезвычайно высокой скорости расщепления полисахарида и, следовательно, возможности почти мгновенной мобилизации заключенных в нем энергетических запасов. Наиболее богаты гликогеном печень и мышцы. Целлюлоза — широко распространенный в природе полисахарид, являющийся составной частью оболочек растительных клеток. в состав древесины входит от 50 до 70 %, а в состав хлопка — до 98 % целлюлозы. Молекула целлюлозы представляет собой линейную цепь, состоящую из остатков D-глюкопиранозы, связанных между собой бета-1,4-гликозидной связью. Молекулярная масса целлюлозы колеблется от 250 000 до 1 000 000 при содержании не менее 1500 остатков глюкозы. Целлюлоза не растворяется в воде и обычных органических растворителях, но растворяется в аммиачном растворе меди (II) гидроксида (реактив Швейцера) и концентрированном растворе цинка хлорида. Гидролиз целлюлозы осуществляется при нагревании в присутствии серной кислоты. Человек и высшие животные не имеют фермента, гидролизующего бета-гликозидные связи целлюлозы, но она является необходимым балластным компонентом пищи, улучшающим пищеварение. Молекула целлюлозы имеет строго упорядоченную конформацию «жесткий стержень», в которой глюкопиранозные остатки расположены линейно. Такое расположение остатков в пространстве обусловлено тем, что гликозидный атом кислорода и атом кислорода при С-4 связаны с пиранозным циклом экваториально. Линейная конформация молекулы закрепляется внутримолекулярными водородными связями. Параллельно уложенные цепи полисахарида удерживаются за счет образования межмолекулярных водородных связей. благодаря такому строению целлюлоза химически сравнительно инертна (нерастворима в воде, с трудом гидролизуется) и обладает высокой механической прочностью. Важное практическое значение имеют производные целлюлозы, наличие трех свободных спиртовых групп в каждом глюкозидном остатке целлюлозы дает возможность получать ее сложные эфиры. так, при обработке целлюлозы смесью азотной и серной кислот образуются нитраты целлюлозы. Свойства и возможности применения этих продуктов зависят от степени нитрования. смесь моно- и динитратов называют коллодийной ватой, или коллоксилином. ее используют для изготовления коллодия, применяющегося в медицине для фиксации повязок. Продукт полного нитрования целлюлозы (целлюлозы тринитрат, тринитроклетчатка, пироксилин) является взрывчатым веществом, используемым в производстве бездымного пороха. большое народнохозяйственное значение имеет целлюлозы диацетат, используемый в производстве ацетатного шелка, а также целлюлозы ксантогенат, применяемый для получения вискозного волокна и целлофана. натриевая соль карбоксиметилцеллюлозы применяется в производстве лекарственных препаратов. Хитин (C8H13NO5)n — природное соединение из группы азотсодержащих полисахаридов, полимер из остатков N-ацетилглюкозамина, связанных между собой β-(1→4)-гликозидными связям. Основной компонент экзоскелета (кутикулы) членистоногих и ряда других беспозвоночных животных, входит в состав клеточных стенок грибов, ряда бактерий и сине-зелёных водорослей, являясь в них аналогом целлюлозы. Распространённость хитина в природе — на втором месте среди биополимеров после целлюлозы. Представляет собой твёрдое бесцветное либо полупрозрачное вещество (жёсткое на ощупь), не растворимое в воде и полярных органических растворителях (этаноле, диэтиловом эфире, ацетоне), растворяется в растворе хлорида лития в диметилацетамиде (при отсутствии следов воды), в концентрированных растворах некоторых солей (хлорид цинка, тиоцианат лития, соли кальция) и в ионных жидкостях. При нагревании с концентрированными растворами минеральных кислот (соляной или серной) происходит гидролиз, в результате образуются мономеры N-Ацетилглюкозамина. При длительном нагревании хитина с концентрированными растворами щелочей происходит N-деацетилирование и образуется хитозан.
Гидроксикарбоновые кислоты: молочная, яблочная, лимонная, винные кислоты. Стереохимия a-гидроксикарбоновых кислот. Лактиды. Лактоны. Фенолокарбоновые кислоты. Салициловая кислота и ее производные. Ацетилсалициловая кислота.
1) Молочная кислота: CH3 – CH – COOH
|
OH
Молочная кислота (лактат) — α-оксипропионовая (2-гидроксипропановая) кислота.
tпл 25—26 °C оптически активная + или — форма.
tпл 18 °C рацемическая форма.
Молочная кислота образуется при молочнокислом брожении сахаров, в частности в прокисшем молоке, при брожении вина и пива.
Соли Лактаты.
Молочная кислота формируется при распаде глюкозы. Иногда называемая «кровяным сахаром», глюкоза является главным источником углеводов в нашем организме. Это основное топливо для мозга и нервной системы, так же как и для мышц во время физической нагрузки. Когда расщепляется глюкоза, клетки производят АТФ (аденозинатрифосфат), который обеспечивает энергией большинство химических реакций в организме. Уровень АТФ определяет, как быстро и как долго наши мышцы смогут сокращаться при физической нагрузке.
Производство молочной кислоты не требует присутствия кислорода, поэтому этот процесс часто называют «анаэробным метаболизмом». Многие считают, что мышцы производят молочную кислоту, когда недополучают кислород из крови. Другими словами, вы находитесь в анаэробном состоянии. Однако учёные утверждают, что молочная кислота образуется и в мышцах, получающих достаточно кислорода. Увеличение количества молочной кислоты в кровотоке свидетельствует лишь о том, что уровень её поступления превышает уровень удаления. Резкое увеличение (в 2-3 раза) уровня лактата в сыворотке крови наблюдается при тяжелых расстройствах кровообращения, таких как геморрагический шок, острая левожелудочковая недостаточность и др., когда одновременно страдает и поступление кислорода в ткани и печеночный кровоток.
Зависимое от лактата производство АТФ очень незначительно, но имеет большую скорость. Это обстоятельство делает идеальным его использование в качестве топлива, когда нагрузка превышает 50 % от максимальной. При отдыхе и умеренной нагрузке организм предпочитает расщеплять жиры для получения энергии. При нагрузках в 50 % от максимума (порог интенсивности для большинства тренировочных программ) организм перестраивается на преимущественное потребление углеводов. Чем больше углеводов вы используете в качестве топлива, тем больше производство молочной кислоты.
2) Яблочная кислота: HOOC – CH – CH2COOH Соли: малаты.
|
OH
Яблочная кислота (оксиянтарная кислота, гидроксибутандиовая кислота)— двухосновная оксикарбоновая кислота. Бесцветные гигроскопичные кристаллы, хорошо растворимые в воде и этиловом спирте. Яблочная кислота содержится в незрелых яблоках, винограде, рябине, барбарисе, малине и др. Растения махорки и табака содержат её в виде солей никотина. Малат является промежуточным продуктом цикла трикарбоновых кислот и глиоксилатного цикла. В цикле Кребса L-яблочная кислота образуется путём гидратации фумаровой кислоты и далее окисляется коферментом НАД+ в щавелевоуксусную кислоту.
3) Винная кислота.
Винная кислота (диоксиянтарная кислота, 2,3-дигидроксибутандиовая кислота) НООС-СН(ОН)-СН(ОН)-СООН — двухосновная оксикислота. Соли тартраты.
Винная кислота — распространённое природное соединение. В значительном количестве она содержится в кислом соке многих фруктов, например, в виноградном соке. D-винную кислоту получают действием минеральных кислот на ее кислую К-соль (винный камень), образующуюся при брожении виноградного сока. При пиролизе D-винная кислота декарбоксилируется с образованием пировиноградной СН3СОСООН и пировинной (метилянтарной) НООССН(СН3)СН2СООН кислот. Она восстанавливается до янтарной кислоты, восстанавливает аммиачный раствор AgNO3 до Ag; в щелочной среде растворяет Сu(ОН)2 с образованием прозрачного ярко-синего раствора - реактива Фелинга.
4) Лимонная кислота.
Лимонная кислота (2-гидрокси-1,2,3-пропантрикарбоновая кислота, 3-гидрокси-3-карбоксипентандиовая)
OH
|
HOOC – CH2 –C – CH2 – COOH — кристаллическое вещество белого цвета, температура плавления
|
HOOC
153 °C, хорошо растворима в воде, растворима в этиловом спирте, малорастворима в диэтиловом эфире. Слабая трёхосновная кислота. Соли и эфиры лимонной кислоты называются цитратами.
Лимонная кислота, являясь главным промежуточным продуктом метаболического цикла трикарбоновых кислот, играет важную роль в системе биохимических реакций клеточного дыхания множества организмов. Сама кислота, как и её соли (цитрат натрия, цитрат калия, цитрат кальция), широко используется как вкусовая добавка, регулятор кислотности и консервант в пищевой промышленности, для производства напитков, сухих шипучих напитков.
Применяется в медицине, в том числе в составе средств, улучшающих энергетический обмен (в цикле Кребса).
В косметике используется как регулятор кислотности, буфер, хелатирующий агент, для шипучих композиций (ванны).
В нефтяной промышленности при бурении нефтяных и газовых скважин используется для нейтрализации цемента в растворе (например, после срезки с цементного моста). Лимонная кислота удаляет ионы кальция из бурового раствора.
При приёме внутрь в небольших дозах (например, при употреблении цитрусовых) активирует цикл Кребса, что способствует ускорению метаболизма. При похмелье рассматривается токсикологами как мера химической дезинтоксикации. Сухая лимонная кислота и её концентрированные растворы при попадании в глаза вызывают сильное раздражение, при контакте с кожей возможно слабое раздражение. При единовременном употреблении внутрь больших количеств лимонной кислоты возможны: раздражение слизистой оболочки желудка, кашель, боль, кровавая рвота. При вдыхании сухой лимонной кислоты — раздражение дыхательных путей.
Классификация аминокислот. Основные Представители природных a-аминокислот, их стереохимия.
Аминокислотами называют
производные карбоновых кислот, в
углеводородном радикале которых один
или несколько атомов водорода замещены
на аминогруппу. В зависимости от природы
углеводородного радикала, с которым
связана карбоксильная группа,
аминокислоты подразделяют на
алифатические и ароматические.
Алифатические аминокислоты по взаимному
расположению аминогруппы и карбоксильной
группы подразделяют на α-, β-, γ- и так
далее аминокислоты. Наиболее
распространенными в природе являются
α-аминокислоты, входящие в состав белков.
Наибольшее значение в химии
имеют α-аминокислоты,
в основном потому, что они являются
мономерами белков. В номенклатуре
α-аминокислот чаще применяют тривиальные
названия: глицин, аланин, валин, лейцин
и др. В биохимии используют также
трехбуквенные сокращения тривиальных
названий: глицин — Гли, аланин — Ала,
валин — Вали др. Систематические названия
для природных аминокислот практически
не применяют. По химической природе
остатка, связанного с α-аминокислотным
фрагментом —CH(NH2)COOH, α-аминокислоты
делятся на алифатические, ароматические
и гетероциклические. В гетероциклических
α-аминокислотах пролине и оксипролине
α-аминокислотный фрагмент входит в
состав гетероцикла:  Все
α-аминокислоты, исключая глицин, содержат
хиральный α-углеродный атом и существуют
в виде пары энантиомеров. для обозначения
конфигураций при хиральном центре
применяют D,L-систему.
Все
α-аминокислоты, исключая глицин, содержат
хиральный α-углеродный атом и существуют
в виде пары энантиомеров. для обозначения
конфигураций при хиральном центре
применяют D,L-систему.
 α-аминокислоты,
входящие в состав белков животных и
человека, имеют L-конфигурацию, аминокислоты
D-ряда встречаются лишь в небелковых
компонентах растений и грибов, а также
синтезируются микроорганизмами.
Некоторые аминокислоты, встречающиеся
в белках, содержат по два хиральных
центра и могут существовать в виде двух
пар энантиомеров. Использование
аминокислот L-ряда для биосинтеза белков
имеет важнейшее значение в формировании
их пространственной структуры и
проявлении биологической активности.
α-аминокислоты,
входящие в состав белков животных и
человека, имеют L-конфигурацию, аминокислоты
D-ряда встречаются лишь в небелковых
компонентах растений и грибов, а также
синтезируются микроорганизмами.
Некоторые аминокислоты, встречающиеся
в белках, содержат по два хиральных
центра и могут существовать в виде двух
пар энантиомеров. Использование
аминокислот L-ряда для биосинтеза белков
имеет важнейшее значение в формировании
их пространственной структуры и
проявлении биологической активности.
Аминокислоты классифицируют по разным критериям:
По строению бокового радикала (функциональным группам):
алифатические (глицин, аланин, валин, изолейцин, лейцин);
ароматические (фенилаланин, тирозин, триптофан);
гетероциклические (триптофан, гистидин, пролин).