

1. Каталитическое гидрирование
Алкины могут быть гидрированы до алкенов или алканов в присутствии катализаторов, таких как палладий на угле (Pd/C) или платина на угле (Pt/C). Гидрирование происходит за счет добавления водорода по тройной связи, причем сначала образуется (Z)-алкен, а затем алкан.

2. Восстановление натрием в жидком аммиаке
Восстановление алкинов натрием в жидком аммиаке приводит к образованию транс-алкенов.
При восстановлении алкинов с помощью натрия в жидком аммиаке р. Берча водород также присоединяется селективно, но с образованием транс-изомера:

3. Использование в синтезе (z)- и (e)-алкенов
Алкины можно использовать как исходные соединения для синтеза (Z)- и (E)-алкенов.
Для получения (Z)-алкенов алкины гидрируют в присутствии катализатора Линдлара (Pd/CaCO3 с добавлением хинолина), который селективно гидрирует тройную связь до (Z)-алкена.
Для получения (E)-алкенов алкины восстанавливают натрием в жидком аммиаке (реакция Берча), а затем протонируют, что приводит к образованию транс-алкена.
Химические свойства алкинов: реакции электрофильного присоединения к тройной связи – галогенирование, гидрогалогенирование, гидратация (реакция Кучерова).
Алкины — непредельные углеводороды, в молекулах которых есть одна тройная связь.
Механизм реакции аналогичен галогенированию алкенов
Реакции присоединения к алкинам:
Галогенирование. Присоединение галогенов к алкинам происходит даже при комнатной температуре в растворе (растворители — вода, CCl4). Это качественная реакция на тройную связь.
Гидрогалогенирование. Алкины присоединяют галогеноводороды. Реакция протекает по механизму электрофильного присоединения с образованием галогенопроизводного алкена или дигалогеналкана.
Гидратация (реакция Кучерова). Присоединение воды протекает в присутствии кислоты и катализатора (соли ртути II). Сначала образуется неустойчивый алкеновый спирт, который затем изомеризуется в альдегид или кетон. Например, при взаимодействии ацетилена с водой в присутствии сульфата ртути образуется уксусный альдегид.
Кислотность ацетилена и терминальных алкинов. Димеризация, тримеризация ацетилена. Полиацетилен.
Благодаря высокой электроотрицательности атома углерода в sр-гибридизации алкины с концевой тройной связью обладают слабой СН-кислотностью и способны замещать атом водорода на металлы и другие группы.
Ацетилен в присутствии меди (I) хлорида и аммония хлорида димеризуется с образованием винилацетилена:

Реакция имеет важное промышленное значение, так как винилацетилен используют в качестве промежуточного продукта при производстве синтетических каучуков. Ацетилен и его гомологи подвергаются циклотримеризации. При нагревании ацетилена в присутствии активированного угля образуется бензол.

Алкины при нагревании в присутствии комплексных никельорганических катализаторов Ni(CO)2[(C6H5)3P]2 подвергаются циклотримеризации с образованием бензола и его замещенных:
Полиацетилен — органическое вещество, полимер ацетилена. Известны цис- и транс-формы полиацетилена. При нагревании цис-изомера до 100-150 °C, совершается переход в транс-форму. В зависимость от способа получения полиацетилен может представлять собой чёрный порошок, сероватый пористый материал, серебристые или золотистые плёнки. Не растворим ни в одном из известных органических растворителей.
применения.
Классификация, номенклатура, структурная изомерия и пространственное строение циклоалканов.
Циклоалканы — это предельные (насыщенные) углеводороды, которые содержат замкнутый углеродный цикл. Общая формула циклоалканов — CnH2n, где n≥3.
Классификация циклоалканов:
малые циклы (3–4 атома углерода);
средние циклы (5–7 атомов углерода);
макроциклы (8 атомов углерода и более).
Номенклатура циклоалканов: названия образуются с помощью добавления приставки цикло- к названию алкана с соответствующим числом атомов углерода. В качестве основной цепи выбирают цикл, нумерацию проводят таким образом, чтобы заместители получили наименьшие номера. Если заместитель один, то его номер не указывается.
Изомерия циклоалканов:
Структурная изомерия: связана с разным числом углеродных атомов в кольце, разным числом углеродных атомов в заместителях и с положением заместителей в цикле.
Межклассовая изомерия: циклоалканы изомерны алкенам.
Пространственная (геометрическая) изомерия: у циклоалканов с двумя заместителями, расположенными у соседних атомов углерода в цикле, заместители могут располагаться по одну сторону от цикла (цис-изомер) или по разные стороны (транс-изомер).
Конформации Циклогексана и его производных, пространственная изомерия производных циклогексана. Типы напряжений в молекулах циклоалканов. Химические свойства циклоалканов (циклобутана, циклопентана и циклогексана, циклопропана).
Конформация циклогексана — одна из нескольких трёхмерных форм, принимаемой молекулой циклогексана.
Основные конформации:
Кресло — наиболее стабильная конформация. При 25 °C почти все молекулы в растворе циклогексана принимают эту конформацию.
Полу-кресло.
Лодка.
Твист.
Для монозамещённых циклогексанов имеются две неэквивалентные конформации с заместителем в аксиальном и экваториальном положениях. Обычно более устойчива экваториальная форма.
Молекула может легко менять конформации, и только две из них — кресло и твист — могут быть изолированы в чистом виде.

Пространственная изомерия производных циклогексана может проявляться в виде:
Цис-транс-изомерии, обусловленной различным взаимным расположением в пространстве заместителей относительно плоскости цикла. В цис-изомерах заместители находятся по одну сторону от плоскости кольца, в транс-изомерах — по разные.
Оптической (зеркальной) изомерии некоторых ди- (и более) замещённых циклов.
Поворотной (конформационной) изомерии циклоалканов. Все циклы, кроме циклопропана, имеют неплоское строение, что обусловлено стремлением атомов углерода к образованию нормальных (тетраэдрических) углов между связями. Это достигается поворотами по σ-связям С–С, входящим в цикл. При этом возникают различные конформации (поворотные изомеры) с разной энергией, и чаще реализуются те из них, которые обладают наименьшей энергией, то есть более устойчивые. Например, в циклогексане наиболее устойчивой является конформация «кресла».
В молекулах циклоалканов могут возникать следующие типы напряжений:
Угловое напряжение (напряжение Байера) — увеличение энергии молекулы, вызванное отклонением угла между связями от величины нормального тетраэдрического угла (109о28′).
Торсионное напряжение (напряжение Питцера, напряжение заслонённых связей) — увеличение энергии, вызванное отклонением конформации любого этанподобного звена в молекуле циклоалкана от заторможенной.
Трансаннулярное напряжение (напряжение Прелога) — увеличение энергии молекулы вследствие взаимодействия несвязанных атомов и фрагментов (двойных связей, функциональных групп и т. д.).
На основании величин общей энергии напряжения все циклоалканы можно разделить на четыре группы:
малые циклы C3 и C4;
нормальные циклы C5–C7;
«средние циклы» C8–C11;
макроциклы с числом атомов углерода большим, чем C11.
Химические свойства циклоалканов зависят от величины цикла. Для малых циклов (циклопропан, циклобутан) характерны реакции присоединения, протекающие с разрывом цикла. Для средних циклов (циклопентан, циклогексан) характерны реакции замещения, протекающие без разрушения цикла.
Примеры химических свойств:
Галогенирование. Циклопропан и циклобутан присоединяют бром, но реакция протекает труднее, чем с пропеном или бутеном.
Гидрирование. Для циклопропана и циклобутана характерно присоединение водорода с образованием соответствующих нормальных алканов. Реакция протекает при нагревании в присутствии катализатора.
Гидрогалогенирование. Малые циклы присоединяют галогеноводороды, образуя галогеналканы.
Дегидрирование. Циклогексан в присутствии катализатора превращается в бензол.
Окисление. Как средние, так и малые циклы окисляются под действием сильных окислителей (например, 50 %-й азотной кислоты). Реакция сопровождается разрывом цикла и образованием дикарбоновых кислот. Например, циклогексан в присутствии катализатора образует адипиновую кислоту, которая используется в производстве синтетических волокон.
Циклоалканы подвергаются полному окислению, сгорая в кислороде с образованием углекислого газа и воды.
Классификация и номенклатура аренов. Природа связей в молекуле бензола. Конденсированные ароматические углеводороды: нафталин, антрацен, фенантрен, бензпирен.
Арены — ароматические углеводороды, содержащие одно или несколько бензольных колец. Общая формула гомологического ряда аренов — CnH2n–6 при n ≥ 6.
Классификация аренов:
В зависимости от числа ароматических колец:
одноядерные арены (бензол и его производные);
полиядерные арены (дифенил, дифенил метан, трифенилметан, нафталин, антрацен и т. д.).
Номенклатура аренов:
При составлении названия ароматического соединения за главную цепь принимают молекулу бензола.
Если в ароматическом кольце несколько заместителей, то атомы углерода бензольного кольца нумеруются в направлении, где больше заместителей, от самого главного заместителя (чем больше атомов углерода в радикале, тем он старше).
Если в молекуле бензола присутствуют два заместителя, то также используют систему специальных приставок:
орто— (о-) — если заместители расположены у соседних атомов углерода в бензольном кольце (1,2-положения);
мета— (м-) — заместители расположены через один атом углерода (1,3-положения);
пара— (п-) — заместители расположены на противоположных сторонах кольца (1,4-положения).
Многие арены имеют свои исторические названия. Например: метилбензол (толуол), изопропилбензол (кумол), винилбензол (стирол).
Молекула бензола представляет собой плоский шестиугольный цикл. Каждый из атомов углерода находится в состоянии sp2-гибридизации. Гибридные орбитали каждого атома участвуют в образовании трёх σ-связей: двух с соседними атомами углерода и одной — с атомом водорода. Угол между этими связями составляет 120°.
Одна p-орбиталь каждого атома углерода остаётся негибридной. За счёт этих орбиталей образуется общая π-электронная система, состоящая из шести электронов.
Все связи между атомами углерода в молекуле бензола одинаковой длины (0,140 нм), что соответствует промежуточному значению между одинарной и двойной (полуторная связь). Соответственно, в молекуле бензола между углеродными атомами нет обычных одинарных и двойных связей, а все они выравнены (делокализованы).
Современное представление об электронной природе связей в бензоле основывается на гипотезе Лайнуса Полинга, который предложил изображать молекулу бензола в виде шестиугольника с вписанной окружностью, подчёркивая тем самым отсутствие фиксированных двойных связей и наличие единого электронного облака, охватывающего все шесть атомов углерода цикла.
Конденсированные ароматические углеводороды — это полициклические арены, молекулы которых содержат не менее двух бензольных колец и имеют общие атомы углерода.
Примеры конденсированных ароматических углеводородов:
Нафталин — состоит из двух конденсированных бензольных колец.
Антрацен — линейно сочленённый изомер.
Фенантрен — ангулярно сочленённый (от лат. angulus — угол, угловой).
Пирен (бензпирен) — периконденсированный углеводород.
Ароматичность, критерии ароматичности. Правило Хюккеля.
Совокупность
специфических свойств бензола, а именно
высокая стабильность, инертность в
реакциях присоединения и склонность к
реакциям замещения, получила общее
название «ароматичность», или
«ароматические свойства». В 1931 году
немецкий ученый Эрих Хюккель на основе
квантово-химических расчетов с помощью
метода МО сформулировал правило
стабильности циклических сопряженных
систем, которое представляет собой
теоретически обоснованный метод,
позволяющий предсказать, будет ли
циклическая сопряженная система
ароматической или нет.
Согласно правилу Хюккеля критерием
ароматичности органического соединения
является наличие в его структуре плоского
цикла, содержащего замкнутую сопряженную
систему, включающую (4n + 2) π-электронов,
где n = 0,1,2,3 и т. д.

Критериями ароматичности являются структурный (выравнивание связей), энергетический (термодинамическая устойчивость), магнитный (наличие кольцевого тока), химический (способность к реакциям электрофильного замещения) факторы.
В связи с особенностями электронного строения бензольного кольца (наличие электронного облака над и под плоскостью цикла) типичными для аренов являются реакции, в которых они служат донорами электронов, основаниями Льюиса, способными реагировать с акцепторами электронов.
Реакции электрофильного замещения в бензоле (галогенирование, нитрование, сульфирование, алкилирование, ацилирование). Представление о механизме реакций электрофильного замещения в ароматическом ряду (- и -комплексы).
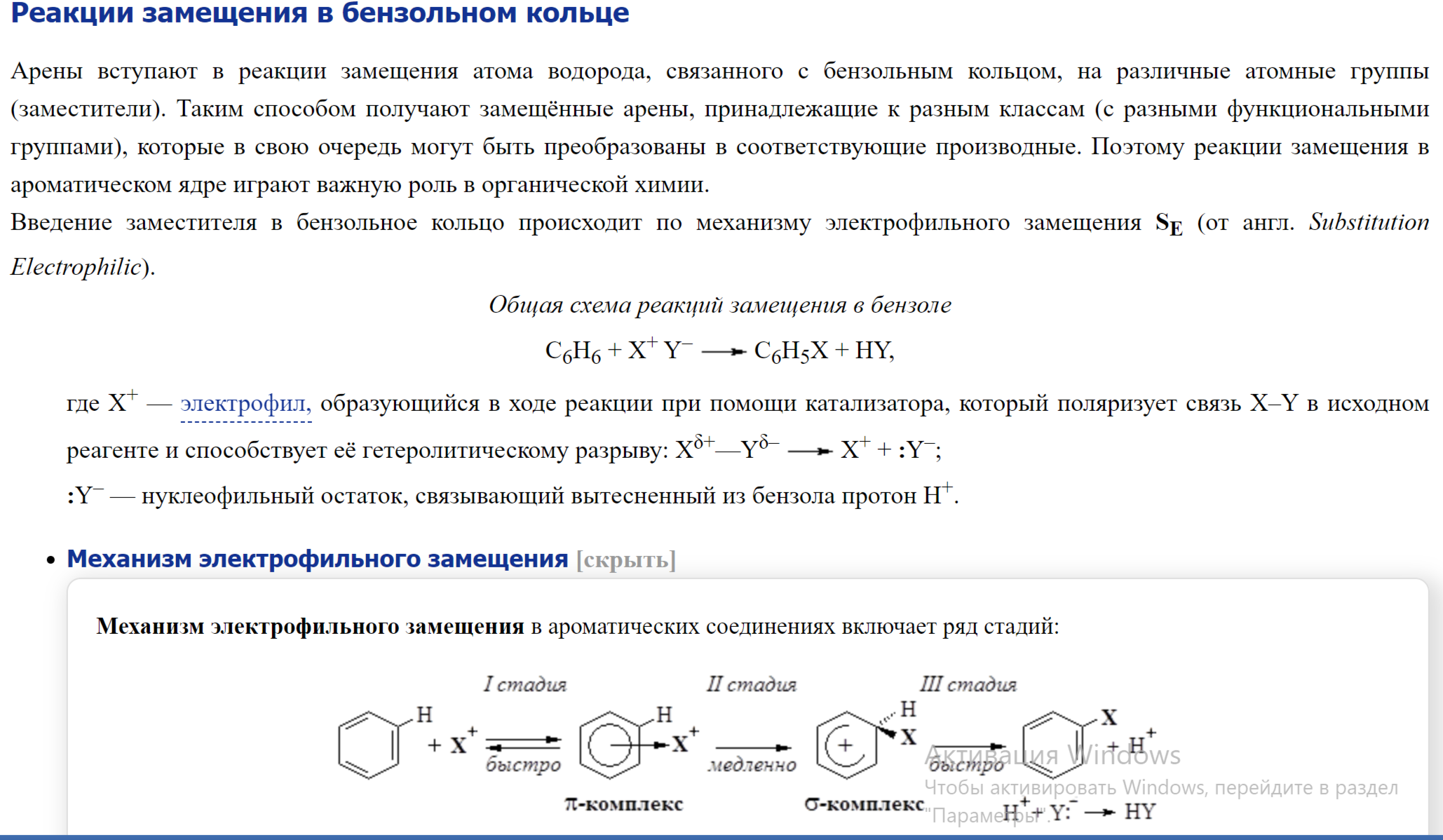


Влияние заместителей в бензольном кольце на изомерный состав продуктов и скорость реакции. Активирующие и дезактивирующие заместители. Орто-, пара- и мета-ориентанты.
В молекуле незамещенного бензола электронная плотность распределена равномерно, поэтому электрофильный реагент может атаковать в равной степени любой из шести атомов углерода. Если в бензольном кольце содержится какой-либо заместитель, то под его влиянием происходит перераспределение π-электронной плотности цикла и новая группа вступает уже в определенные положения по отношению к имеющемуся заместителю. В реакциях электрофильного замещения монозамещенных бензола, в зависимости от электронной природы заместителя, вступающая группа может занимать преимущественно орто-, мета- или пара-положения, а реакция соответственно протекать быстрее или медленнее, чем с незамещенным бензолом. По влиянию на направление реакций электрофильного замещения и реакционную способность бензольного кольца заместители можно разделить на две группы — заместители I рода (орто-, пара-ориентанты) и заместители II рода (мета-ориентанты). К заместителям I рода относятся атомы и атомные группы, проявляющие положительный индуктивный или положительный мезомерный эффекты: —O–, —NR2, —NHR, —NH2, —OH, —OR, —NHCOR, —OCOR, —SR, —F, —Cl, —Br, —I, —Alk и др. Заместители I рода (за исключением галогенов) увеличивают электронную плотность в бензольном кольце и тем самым активируют его в реакциях электрофильного замещения.
К заместителям II рода относятся группы, проявляющие отрицательный индуктивный или отрицательный мезомерный эффекты: —NO2, —SO3H, —CN, —CHO, —COR, —COOH, —COOR, —CONH2, —CCl3 и др. Заместители II рода уменьшают электронную плотность в бензольном кольце и снижают скорость реакции электрофильного замещения по сравнению с незамещенным бензолом.
Механизм влияния заместителей в бензольном кольце на направление и скорость реакций электрофильного замещения можно объяснить с учетом электронных эффектов, которые играют существенную роль как в распределении электронной плотности в стационарном состоянии молекулы (статический фактор), так и в стабилизации образующихся в процессе реакции σ-комплексов (динамический фактор). Заместители I рода (кроме галогенов) за счет +I-или +М-эффекта проявляют электронодонорные свойства. Они повышают электронную плотность на всех атомах углерода бензольного кольца, но в большей степени на углеродных атомах в орто- и пара-положениях (статический фактор). Это является причиной облегчения электрофильного замещения в сравнении с реакциями SE у незамещенного бензола и преимущественной атаки электрофильной частицей орто- и пара-положений. Заместители II рода, наоборот, за счет –I- или –М-эффектов проявляют электроноакцепторные свойства, вызывая общее уменьшение электронной плотности в бензольном кольце, но в большей степени это влияние сказывается в ортои пара-положениях. Поэтому они затрудняют реакции электрофильного замещения вообще и особенно с участием орто- и пара-положений. В результате замещение протекает преимущественно в мета-положении.
Кроме заместителей I и II рода, имеется небольшое число заместителей, проявляющих смешанное действие (—CH2NO2, —CH2Hal, —CH2OH, —CHHal2 и др.). Эти заместители несколько затрудняют электрофильное замещение в бензольном ядре, но в результате реакции, как правило, образуется смесь примерно равных количеств орто-, мета- и пара-изомеров.
Реакции радикального замещения и окисления в боковой цепи. Причины устойчивости бензильных радикалов.
Алкилбензолы, в отличие от незамещенного бензола, окисляются значительно легче. При действии сильных окислителей (KMnO4, K2Cr2O7 и др.) окислению подвергаются боковые углеродные цепи. Продуктами окисления являются ароматические карбоновые кислоты. Причем каждый алкильный радикал в бензольном кольце, независимо от длины углеродной цепи, окисляется в карбоксильную группу.

Окисление алкилбензолов является важным способом получения ароматических карбоновых кислот. Взаимодействие гомологов бензола с галогенами (хлором или бромом) в условиях свободнорадикального замещения осуществляется с участием боковой цепи. При этом на атом галогена замещается, как правило, атом водорода при атоме углерода, непосредственно связанном с бензольным кольцом (α-положение):

такое направление замещения обусловлено образованием в качестве промежуточной активной частицы свободного радикала бензильного типа, в котором электронная плотность значительно делокализована за счет сопряжения с бензольным кольцом.
Стабильность бензильного радикала обусловлена делокализацией неспаренного электрона за счёт взаимодействия с бензольным кольцом.
Классификация, номенклатура, изомерия галогенуглеводородов.
Галогенуглеводороды — это органические вещества, содержащие в молекуле атомы галогена (чаще брома и хлора).
Классификация:
По числу атомов галогена в молекуле: моногалогеналканы, дигалогеналканы и т. д., также используют общий термин полигалогеналканы для соединений с несколькими галогенами.
По наличию или отсутствию кратных связей: предельные (галогеналканы) и непредельные галогенуглеводороды. Среди непредельных особо выделяются винилгалогениды — это галогеналкены, у которых галоген расположен непосредственно при двойной связи.
Ароматические галогенуглеводороды (галогенопроизводные аренов).
Номенклатура: согласно правилам ИЮПАК галогенуглеводороды рассматриваются как продукты замещения углеводородов, соответствующие атомы галогенов указываются в префиксе. Если это необходимо, положение атома галогена указывается цифрой.
Изомерия:
изомерия углеродного скелета (нормальная и разветвлённая цепь);
изомерия расположения атомов галогенов в молекуле;
оптическая изомерия, связанная с хиральностью атома углерода.
Реакции нуклеофильного замещения атома галогена, их использование в синтезе органических соединений различных классов (спиртов, простых и сложных эфиров, аминов, тиолов и сульфидов, нитроалканов, нитрилов). Представление об идеализированных механизмах SN1 и SN2.
Реакции нуклеофильного замещения — это тип органической реакции, при которой нуклеофил (донор эл. Пары, т.е. кис-ты Льюиса) вступает в реакцию с субстратом путём замены уходящей группы.
С помощью реакций нуклеофильного замещения алкилгалогениды можно превратить в:
Спирты. Например, при обработке 2-хлорпропана водным раствором щелочи образуется 2-пропанол.
Простые эфиры. Например, при взаимодействии метилбромида с этилатом натрия образуется метилэтиловый эфир.
Сложные эфиры. Например, при взаимодействии этилбромида с ацетатом натрия образуется этилацетат.
Амины. Например, при обработке алкилгалогенидов водным или спиртовым раствором аммиака образуются амины.
SN1 (мономолекулярное нуклеофильное замещение) — двухстадийный механизм, который включает следующие стадии:
Ионизация субстрата с образованием карбкатиона (медленная стадия).
Нуклеофильная атака карбкатиона (быстрая стадия).
Отщепление катиона (быстрая стадия).
SN1 реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути обычно реагируют только третичные и вторичные алкилпроизводные, а также аллильные и бензильные субстраты.
SN2 (бимолекулярное нуклеофильное замещение) — одностадийный механизм, который заключается в практически одновременном отщеплении галогенид-иона и присоединении гидроксид-аниона (без образования карбокатиона).
Следует помнить, что чистый SN1 или SN2 являются всего лишь идеальными (предельными) модельными случаями, так как один и тот же субстрат может реагировать с одним и тем же нуклеофилом, в зависимости от условий реакции и растворителя, как по механизму SN1, так и SN2.
Соединения с повышенной подвижностью атома галогена. (аллил- и бензилгалогениды). Соединения с пониженной подвижностью атома галогена (винилхлорид и хлорбензол). Реакции элиминирования галогеноводорода. Правило Зайцева.
Аллил- и бензилгалогениды — это соединения с повышенной подвижностью атома галогена в сравнении с насыщенными галогеналканами.
Химические свойства:
очень легко вступают в реакции нуклеофильного замещения. Например, гидролиз аллил- и бензилбромидов осуществляется кипячением с водой, а при использовании водного раствора NaOH (более сильного нуклеофила) реакция происходит уже при комнатной температуре;
могут реагировать и по механизму SN2, если созданы соответствующие условия (высокая концентрация нуклеофила, малополярный слабоионизирующий растворитель).
Причина повышенной реакционной способности: пониженная энергия диссоциации связи С-Hal из-за стабильности карбокатионов, образующихся после разрыва связи С-Hal.
Соединения с пониженной подвижностью атома галогена — это галогенопроизводные аренов и ароматических углеводородов, в которых атом галогена непосредственно связан с одним из атомов углерода двойной связи.
Примеры:
Винилхлорид (CH₂=CHCl) — простейшее хлорпроизводное этилена. Атом хлора у двойной связи по сравнению с галогеналканами становится значительно менее подвижным.
Хлорбензол — бесцветная, летучая, хорошо растворимая в воде жидкость. Атом галогена замещается с трудом в жёстких условиях.
Низкая подвижность галогена объясняется эффектом сопряжения между кольцом и галогеном, вследствие которого связь углерод–галоген укорачивается и становится более прочной.
Реакции элиминирования — это реакции, при которых происходит отщепление атомов или атомных групп от молекулы исходного вещества при сохранении её углеродного скелета.
К реакциям элиминирования относятся:
Дегидрогалогенирование (дегидрохлорирование — отщепление HCl, дегидробромирование — отщепление HBr и т. д.).
Дегалогенирование (отщепление Hal2).
Дегидратация (отщепление воды).
Дегидрирование (отщепление водорода).
Декарбоксилирование (отщепление СО2).
Декарбонилирование (отщепление СО) и др.
В зависимости от расположения отщепляемых групп в молекулах исходного вещества реакции элиминирования подразделяются на α-, β-, γ-элиминирование и т. д.
Правило Зайцева — в органической химии эмпирическое правило, используемое для предсказания преобладающего продукта в реакциях отщепления воды или галогеноводородов от спиртов и галогенидов соответственно.
Формулируется следующим образом: при дегидратации вторичных и третичных спиртов и при дегидрогалогенировании вторичных и третичных галогенидов водород отщепляется преимущественно от наименее гидрогенизированного атома углерода.
Литий- и магнийорганические соединения и их использование в органическом синтезе.
В органической химии выделяют особую группу соединений – элементорганические соединения. В этих соединениях атом C образует химическую связь с различными другими элементами, не считая атомов H, Hal, O, N и S. Названия МОС образуют от названий углеводородного радикала и металла, например: бутиллитий, диэтилмагний, алкилмагнийхлорид. Литийорганические соединения образуются при взаимодействии металлического лития с RHal в растворителях, которые взаимодействуют с ионом металла (сольватируют металлорганические соединения). Реакцию проводят в инертной атмосфере (N2, аргон), чтобы избежать реакции окисления кислородом воздуха: R–X + 2Li → R–Li + LiX, (X = Cl, Br, I). Связь C–Li – очень полярна. Обычно литийорганические соединения в чистом виде не получают и не используют, так как они энергично реагируют с O2, CO2, H2O и может произойти самовоспламенение. Поэтому их получают в растворах, которые сразу используют в дальнейших реакциях. Литийорганические соединения являются сильнейшими С-нуклеофилами и С-основаниями. Для них характерны реакции с различными кислотами и другими электрофильными реагентами, реагируют с соединениями с подвижным Н – атомом (Н2О, ROH, NH3, первичные (RNH2) и вторичные (R2NH) амины и др.). В основном реакции литийалкилов протекают таким же образом, как и для соответствующих магнийорганических соединений, например:

т. е. имеет место присоединение углеводородного радикала к полярной С=О связи. В некоторых случаях присоединение происходит и к малополярным или неполярным двойным С=С–связям.
При окислении литийорганических соединений кислородом воздуха в растворе конечными продуктами являются спирты (фенолы в случае Ar–Li) в виде их Li-солей. Промежуточными продуктами являются гидропероксиды и их соли:

Литийорганические соединения широко используются в органическом синтезе в качестве промежуточных продуктов. Например, их применяют для промышленного синтеза комплексных металлорганических катализаторов с целью проведения стереорегулярной полимеризации алкенов, алкадиенов, алкинов.
Магнийорганические соединения бывают 2-х типов: с одним углеводородным остатком – RMgХ, и двумя – R2Mg (диалкилмагний). Магнийорганические соединения получают прямым взаимодействием Mg (в виде стружки) с RHal обычно в растворе сухого диэтилового эфира или ТГФ:
![]()
Эту реакцию открыл французский химик Гриньяр (1901г.), а магнийорганические соединения часто называют реактивами Гриньяра. Доказано, что структура магнийорганических соединений включает координационно-связанные с атомом Mg молекулы растворителя – диэтилового эфира. Это обеспечивает растворимость реактивов Гриньяра, они прочно удерживают молекулы растворителя и при его удалении обычно разрушаются.. Независимо от строения соединения в растворе пользуются обычно простой формулой RMgХ. Свойства RMgХ в основном подобны свойствам литийорганических соединений, т.е. во многом определяются полярностью связи С–Mg, только активность RMgХ меньше. они легко подвергаются атаке со стороны электрофильных реагентов. Это и определяет свойства магнийорганических соединений как восстановителей, оснований и нуклеофилов. При обработке RMgХ галогенидами металлов-окислителей, которые не образуют прочной связи с углеродом, образуются алканы. Галогениды металлов, способные восстанавливаться под действием магнийорганических соединений и затем образовывать достаточно прочную связь с углеродом, используются для синтеза других элементоорганических соединений. Вода, спирты, фенолы (Ar–OH), первичные и вторичные амины, карбоновые кислоты, т.е. соединения с подвижным атомом Н, разлагают реактивы Гриньяра с образованием углеводородов:
![]()
Если в качестве RMgХ брать СН3MgI, то подобные реакции служат для количественного определения активного водорода (метод Чугаева-Церевитинова) по объему выделившегося метана (количество СН4 соответствует количеству, числу подвижных атомов Н):
![]()
Если в реакцию с RMgХ вступает муравьиный альдегид, то образуются первичные спирты:

Если с RMgХ реагируют другие альдегиды – образуются вторичные спирты, а с кетонами – третичные. Реактивы Гриньяра применяются для синтеза 1-алкенов (исходя из аллилгалогенидов):
![]()
Взаимодействие реактивов Гриньяра с α-окисями приводит к спиртам:
![]()
Реакция и с серой:

При получении RMgХ в эфире можно не опасаться его последующего окисления: пары кипящего эфира практически исключают контакт кислорода воздуха с реактивом Гриньяра.
Биологическое действие галогенпроизводных, их применение в народном хозяйстве. Хлороформ, иодоформ, перфторуглеводороды, перфторполиэтилен (тефлон). Инсектициды.
Галогенпроизводные алифатического ряда обладают высокой токсичностью для насекомых, микроорганизмов, растений и животных. С увеличением молекулярной массы галогеналканов токсичность их повышается, за исключением производных метана, которые по токсичности превосходят все другие соединения этого класса. Установлено, что галогеналканы разветвлённого строения менее токсичны, чем галогеналканы той же молекулярной массы с нормальным строением углеродной цепи. С увеличением числа атомов галогена в метилгалогенидах токсичность этих соединений для насекомых понижается, тогда как для производных гомологов метана инсектицидность несколько возрастает. Биологическая активность галогеналканов и галогеналкенов и их токсичность для различных видов живых организмов зависит от строения и реакционной способности соединений. Это свидетельствует о том, что производные алифатических углеводородов взаимодействуют с жизненно важными системами этих организмов. При этом инсектицидная активность галогенпроизводных алкенов значительно выше, чем галогеналканов.
Хлороформ СНCl3. Для его получения берут этиловый спирт СН3—СН2—ОН или ацетон СН3—СО—СН3 и белильную известь. В первом случае, кроме хлороформа, получается муравьинокислый натрий Н—COONa, во втором — уксуснокислый натрий СНз—COONa. Хлороформ получают также непосредственным хлорированием метана в соответствующих условиях. Хлороформ представляет собой нерастворимую в воде жидкость относительной плотности 1,488 (при 20°С) с сильным сладковатым запахом, кипящую при 61,2° С. При вдыхании хлороформ вызывает наркоз, а при более длительном действии — и смерть. Благодаря наркотическому действию он долгое время находил широкое применение при хирургических операциях. Хлороформ хорошо растворяет жиры и другие органические вещества, а потому может применяться как растворитель. В современной промышленности синтетических материалов хлороформ приобрел важное значение как исходный продукт в синтезе ненасыщенных фторуглеродов, полимеризацией которых получают фторкаучуки и фторопласты — материалы, сочетающие высокую термостойкость с очень высокой химической стойкостью. Хлороформ довольно нестоек. При действии света он окисляется воздухом, образуя угольный ангидрид, соляную кислоту, хлор и фосген СОCl2. Последний весьма ядовит. Атомы галоида в хлороформе легко могут замещаться на другие атомы или группы. При действии разбавленной водной щелочи получаются соли муравьиной кислоты. При действии концентрированной щелочи образуется окись углерода. При действии на хлороформ аммиака и едкого кали образуется цианистый калий.
Йодоформ CHJ3 получается из спирта или ацетона при действии иода и едких щелочей или карбонатов щелочных металлов. Более совершенным методом является электролиз йодистого калия или натрия в спиртовом растворе. При электролизе образуются иод и щелочь, необходимые для процесса. Образующийся при реакции йодистый калий (натрий) снова подвергается электролизу, и, таким образом, весь иод йодистой соли идет на образование йодоформа. Йодоформ образует желтые кристаллы с т, пл. 119° С. Это — прекрасный антисептик, применявшийся в хирургии при перевязке ран и пр. Широкому применению йодоформа для этой цели препятствует неприятный навязчивый запах.
Перфторуглеводороды — углеводороды, в которых все атомы водорода замещены на атомы фтора. Фторуглероды отличаются от соответствующих углеводородов большей плотностью и, как правило, более низкими значениями температуры кипения. Высшие и особенно полициклические фторуглероды обладают аномально высокой способностью растворять газы, например, кислород, углекислый газ. Устойчивы к действию кислот, щелочей и окислителей; при нагревании выше 600—800 °C или в условиях радиолиза разлагаются с образованием смеси низших и высших фторуглеродов. По величине температуры кипения для данной молекулярной массы насыщенные фторуглероды близки к благородным газам. В природе фторуглероды не найдены и могут быть получены лишь в результате химического синтеза. Высокая растворимость газов в жидких перфторуглеродах обусловлена наличием в таких жидкостях многочисленных крупноразмерных (в молекулярном масштабе) пустот, в которые способны внедряться молекулы газов. Химически весьма инертны. Не реагируют с кислотами и щелочами даже при нагревании. При нагревании реагируют с щелочными металлами (может быть взрыв). Способны подвергаться пиролизу и фотолизу. Фторуглероды — диэлектрики, теплоносители, гидравлические жидкости, смазочные масла, низкотемпературные хладагенты, мономеры в производстве фторполимеров, эффективные газопереносящие среды, что позволяет использовать их для жидкостного дыхания или в качестве искусственной крови. Конденсация перфторуглеводородов используется для пайки печатных плат. Многие фторуглероды трудногорючи, невзрывоопасны, малотоксичны. Перфторуглеводороды способны создавать сильный парниковый эффект в тысячи раз сильнее, чем CO2, что потенциально может быть использовано для терраформирования.
Тефлон — белое, в тонком слое прозрачное вещество, по виду напоминающее парафин или полиэтилен. Обладает высокой тепло- и морозостойкостью, остаётся гибким и эластичным при температурах от −70 до +270 °C, прекрасный изоляционный материал. Тефлон обладает очень низкими поверхностным натяжением и адгезией и не смачивается ни водой, ни жирами, ни большинством органических растворителей. По своей химической стойкости превосходит все известные синтетические материалы и благородные металлы. Не разрушается под влиянием щелочей, кислот и даже смеси азотной и соляной кислот. Разрушается расплавами щелочных металлов, фтором и трифторидом хлора. В различных отраслях промышленности волокна, полученные из политетрафторэтилена (тефлон, полифен), нашли широкое применение в качестве высокотемпературных мешочных фильтров, разных типов теплостойких прокладок, нитей для текстильных тканей, а также в автомобильном оснащении, промышленных фильтрах общего назначения, элементах запорных и регулирующих клапанов, мешалок и насосов, оборудования для фильтрации и разделения. Благодаря биологической совместимости с организмом человека политетрафторэтилен с успехом применяется для изготовления имплантатов для сердечно-сосудистой и общей хирургии, стоматологии, офтальмологии. Тефлон считается наиболее пригодным материалом для производства искусственных кровеносных сосудов и сердечных стимуляторов. В стоматологии нерезорбируемые мембраны из ПТФЭ с усилением титановым каркасом или без последнего, используются при методиках направленной костной регенерации (НКР). Также существует шовный материал из ПТФЭ. Из-за низкого трения и несмачиваемости насекомые не способны ползти по тефлоновой стене. В частности, тефлоновая защита применяется при содержании нелетающих насекомых, чтобы они не смогли вылезти наружу. Из-за низкой адгезии и хорошей термостойкости тефлон используется в качестве антипригарного покрытия для сковородок и другой посуды. Изделия, в производстве которых используется тефлон: конфорки плит; скалки (с противоналипающим покрытием); кипятильники; воки; бритвенные лезвия. При попадании в организм политетрафторэтилен безвреден.
В группе синтетических контактных инсектицидов по значимости и широте применения до 70-х годов XX столетия выделялись полигалогснопроизводные, из которых 4,4-дихлордифенилтрихлорэтан (ДДТ) и гексахлорциклогексан (ГХЦГ, гексахлоран) были наиболее широко применяемыми препаратами. За открытие действия ДДТ на насекомых П. Мюллер в 1948 г. получил Нобелевскую премию. Оба препарата были доступны и дешевы. ДДТ получали конденсацией хлорбензола с хлоралем под действием концентрированной серной кислоты, а гексахлоран — при фотохимическом хлорировании бензола. Однако большинство полихлоропроизводных — очень устойчивые соединения, крайне медленно разлагающиеся в природных условиях и накапливающиеся в живых организмах. В связи с этим применение ДДТ запрещено. По-видимому, и другие еще используемые пол и галогенопроизводные также будут запрещены к применению в ближайшие годы. Серьезной проблемой при применении инсектицидов стало резистентность насекомых к действию препарата.
Одноатомные спирты. Номенклатура, изомерия. Электронное строение. Физические свойства спиртов, роль водородной связи.
Одноатомными спиртами называют гидроксильные производные углеводородов, содержащие одну гидроксильную группу, связанную с атомом углерода в sp3-гибридизации. Для названия спиртов наиболее часто применяют заместительную и радикало-функциональную номенклатуру IUPAC. По заместительной номенклатуре название спирта образуют из названия углеводорода, соответствующего главной углеродной цепи, к которому прибавляют суффикс -ол с указанием положения гидроксильной группы в цепи углеродных атомов. Нумерацию главной углеродной цепи начинают с того конца, к которому ближе расположена гидроксильная группа. По радикало-функциональной номенклатуре названия спиртов образуют из соответствующего названия углеводородного радикала, связанного с гидроксильной группой, к которому добавляют -овый и спирт. Иногда для названия спиртов используют рациональную номенклатуру, согласно которой спирты рассматривают как производные метилового спирта СН3ОН, получившего название «карбинол». Для некоторых спиртов достаточно распространенными остаются тривиальные названия:

Для спиртов характерна структурная, геометрическая и оптическая изомерия.
Структурная изомерия спиртов обусловлена различным положением гидроксильной группы в углеродной цепи, а также строением углеродного скелета. Для ненасыщенных спиртов структурная изомерия может быть обусловлена и положением кратной связи.
Геометрическая изомерия характерна для непредельных спиртов и определяется различным расположением заместителей относительно плоскости двойной связи.
Оптическая изомерия возможна для спиртов, имеющих в своей структуре асимметрический атом углерода.
Насыщенные одноатомные спирты — бесцветные жидкости или кристаллические вещества со специфическим запахом. Низшие члены гомологического ряда имеют характерный «спиртовый» запах; для бутанолов и пентанолов присущ неприятный «сивушный» запах; высшие алканолы обладают приятным фруктовым запахом. циклоалканолы, непредельные и ароматические спирты в большинстве случаев представляют собой жидкие или твердые вещества, имеющие приятный ароматный запах. так, циклогексанол имеет запах камфоры, пропаргиловый спирт нс3с–сн2–он обладает запахом герани, а 2-фенилэтанол — запахом роз.
Спирты имеют более высокие температуры плавления и кипения, большую растворимость в воде, чем соответствующие углеводороды. такое резкое различие между физическими свойствами спиртов и алканов обусловлено тем, что спирты являются полярными соединениями. они имеют две полярные связи с–о и о–н. существование на атомах гидроксильной группы частичных зарядов противоположного знака приводит к межмолекулярному взаимодействию гидроксильных групп и образованию водородных связей.
В результате такого взаимодействия происходит ассоциация молекул спирта.
Водородные связи значительно слабее ковалентных, однако их образование существенно уменьшает летучесть, повышает температуру кипения, так как образующиеся ассоциаты имеют большую молекулярную массу. Например, этан кипит при –89 °С, тогда как этанол — при 78,5 °С.
Спирты с небольшой молекулярной массой хорошо растворимы в воде. Метанол, этанол, пропанолы, аллиловый и пропаргиловый спирты смешиваются с водой во всех соотношениях. В водных растворах спиртов образуются водородные связи между молекулами воды и спирта.
Образующиеся водородные связи более прочны, чем связи между молекулами спирта, что приводит к уменьшению суммарного объема воды и спирта при смешивании (явление контракции спирта).
Химические свойства спиртов: кислотно-основные свойства. Алкоголяты металлов, их основные и нуклеофильные свойства.
Согласно теории Бренстеда-Лоури спирты — достаточно слабые кислоты. Кислотность спиртов уменьшается по мере усложнения углеродного скелета.
Примеры кислотных свойств спиртов:
Взаимодействие со щелочными металлами. Подобно воде, спирты реагируют с щелочными металлами с образованием алкоголятов и выделением водорода.
Взаимодействие с карбоновыми кислотами. При взаимодействии спиртов с кислотами образуются сложные эфиры.
Основные свойства спиртов:
При действии сильных минеральных кислот спирты могут проявлять свойства оснований.
Таким образом, спирты, проявляя слабые кислотные и слабые основные свойства, являются амфотерными соединениями.
Алкоголяты (алкоксиды) — соединения общей формулы R—OM, где R — алкил (или замещённый алкил), а M — катион металла либо другой катион. Формально — продукты замещения иона водорода гидроксильной группы спиртов другим катионом.
Основные свойства алкоголятов:
белые кристаллические вещества, растворимые в воде;
способны к электролитической диссоциации (в спиртовых растворах они обладают значительной электропроводностью и иногда реагируют в ионной форме);
не растворимы в органических растворителях (за исключением спиртов).
Нуклеофильные свойства алкоголятов:
используются для получения простых эфиров;
способны к образованию «ониевых» соединений с рядом других органических молекул.
Реакции нуклеофильного замещения с участием спиртов. Биологически важные реакции нуклеофильного замещения с участием эфиров фосфорных кислот.
Реакция этерификации – образование сложных эфиров. (нуклеофильные свойства спиртов). Спирты взаимодействуют с минеральными и органическими кислотами с образованием сложных эфиров и воды:

Реакция с ортофосфорной кислотой
 +
3
+
3![]() =
= +
3H2O
+
3H2O
Механизм реакции этерификации.
Образование сложных эфиров при взаимодействии карбоновых кислот со спиртами (этерификация) происходит в условиях кислотного катализа как реакция нуклеофильного замещения.

Стадия I. Активация карбоновой кислоты под действием катализатора – сильной кислоты (например, конц. H2SO4), превращающей нейтральную молекулу в карбокатион.
Стадия II (лимитирующая). Нуклеофильное присоединение спирта к карбокатиону.
Стадия III. Миграция протона H+ и формирование хорошей уходящей группы H2O.
Стадия IV. Отщепление воды и катализатора (H+) от неустойчивого продукта присоединения с образованием cложного эфира.
Процесс этерификации катализируется как кислотами, так и основаниями.
Механизм кислотного катализа заключается в протонировании кислорода карбоксильной группы, что приводит к увеличению электронного дефицита на атоме углерода карбоксильной группы и ускорению атаки молекулой спирта – нуклеофильным реагентом.

Механизм основного катализа заключается в повышении нуклеофильности молекулы спирта по обменной реакции:
