

3 курс / Патологическая физиология / Сахарный диабет
.pdfКафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
-Компенсаторное: высокий уровень глюкозы в крови в некоторой степени обеспечивает ее проникновение в ткани, компенсируя тем самым нарушение транспорта глюкозы клеточной мембраной при инсулиновой недостаточности.
-Патогенетическое - гипергликемия приводит к перегрузке и истощению оставшихся неповрежденных и функционально неполноценных β-клеток, т.е., приводит к усугублению течения диабета. Кроме того,
гипергликемия способствует развитию ангиопатий, нейропатий, катаракты. При очень высоких концентрациях
глюкозы может развыться гипергликемическая (гиперосмолярная) кома.
2. Нарушения белкового обмена. При СД угнетается синтез белков, в результате чего:
-снижается иммунная реактивность организма (недостаточный синтез антител),
-угнетается воспалительный процесс, нарушается заживление дефекта, образуются трофические язвы,
-в условиях детского и юношеского диабета нарушаются процессы роста,
-в условиях гипергликемии в результате неферментативного гликирования белков нарушается их деятельность.
Эти нарушения имеют большое значение в патогенезе развития диабетической ангиопатии и катаракты.
3. Нарушение липидного обмена. В результате недостаточности инсулина угнетается переход жиров в жиро-
вую ткань и в ней усиливается липолиз (расщепление триглицеридов), а также липолитическое воздействие СТГ и АКТГ. В результате увеличивается содержание свободных жирных кислот в крови. Описанными расстройства-
ми обьясняется потеря веса больного, не получающего лечение. Избыточная мобилизация жирных кислот спо-
собствует ресинтезу триглицеридов в печени, что может привести к развитию жировой инфильтрации печени.
Во время диабета снижается активность липопротеиновой липазы, которая гидролизирует триглицериды хиломикронов и β-липопротеидов. Снижение ее активности может быть связано с недостаточностью инсулина и липокаина. Последний продуцируется в мелких протоках эпителия поджелудочной железы и стимулирует синтез фосфолипидов в печени, способствуя метаболизму и удалению жиров из печени, тем самым предотвращая развитие жировой инфильтрации печени.
Выраженные расстройства липидного обмена при СД развиваются тогда, когда недостаточность инсулина осложняется дефицитом липокаина. В этом случае развивается тотальный панкреатический диабет.
4.Глюкозурия и полиурия. При достижении концентрации глюкозы в крови 9-10 ммоль/л (180мг%), она преодолевает почечный барьер, и глюкоза появляется в моче. Повышение содержания глюкозы в первичной моче увеличивает ее осмотическое давление и затрудняет реабсорбцию воды. В результате развивается осмотический диурез и полиурия. Вместе с глюкозой организм теряет значительное количество воды, вследствие чего у больных отмечается постоянное чувство жажды и они ежедневно выпивают до 20 литров воды (полидипсия).
5.Кетонемия и кетоацидоз. Дефицит инсулина при СД приводит к тому, что:
a)Контрисулярные гормоны (адреналин, глюкагон, глюкокортикоиды и т.д.) стимулируют мобилизацию жиров из жировых депо, что является адаптивным механизмом, поскольку ткани обеспечиваются альтернативным субстратом окисления при нарушении утилизации глюкозы.
б) Преобладает воздействие глюкагона, стимулирующего кетогенез в печени.
в) В норме кетоновые тела стимулируют высвобождение инсулина, тем самым подавляя липолиз и кетогенез.
При СД этот регулирующий механизм нарушается: из-за усиленного β-окисления жирных кислот увеличивается образование кетоновых тел в печени.
г) При СД в большом количестве образуется продукт β-окисления жирных кислот - ацетил-CoA, однако его использование в цикле Кребса значительно снижено. В результате избыток ацетил-CoA становится источником образования кетоновых тел. Образовавшиеся β-оксимасляная кислота, ацетоуксусная кислота и ацетон выводятся с мочой в виде солей натрия (кетонурия), а ацетон выводится также легкими. Этот механизм способствует также развитию осмотического диуреза.
11
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
6. Нарушение КОС. В результате накопления кислых продуктов метаболизма развивается метаболический ацидоз.
7. Отрицательный азотистый баланс. Из гюконеогенетические аминокислот усиливается глюконеогенез, что приводит к дефициту свободных аминокислот в тканях, нарушению синтеза белка. Стимулируется синтез мочевины. В результате развивается отрицательный азотистый баланс, то есть когда из организма выводится азота больше, чем поступает.
8. Гиперосмотическая дегидратация тканей. Обусловлена удалением с мочой большого количества гидратных соединений - глюкозы, кетоновых тел, азотсодержащих соединений, Na+, K+, Cl- и других.
12.1.5. ОСЛОЖНЕНИЯ САХАРНОГО ДИАБЕТА
Различают острые и поздние осложнения СД. К острым осложнениям относятся комы. Различают следующие виды комы:
1. Кетоацидотическая кома. У больных, госпитализированных по поводу СД, этот вид комы встречается в 1-
6% случаев. Характерна для тип I СД. Как уже было указано, в печени при недостатке инсулина усиливается синтез кетоновых тел. В результате развивается гиперкетонемия, приводящая к развитию метаболического ацидоза. Метаболический ацидоз приводит к повреждению нейронов, постепенно развивается помутнение, а
потом и потеря сознания (кома). При кетоацидотической коме показано незамедлительное введение инсулина.
2. Гиперосмолярная (гипергликемическая) кома. Характерна для тип II СД. При этом кетоацидоз не развивается. Гипергликемическая кома встречается в 10 раз реже, чем кетоацидотическая. Главным патогенетическим звеном этой комы является выраженная гипергликемия, которая значительно повышает осмотическое давление крови и внеклеточной жидкости, приводя к перемещению жидкости из внутриклеточного пространства во внеклеточное. Дополнительную роль играет гиповолемия, развивающаяся вследствие глюкозурии и полиурии, и приводящая к гипотензии и нарушению перфузии головного мозга.
Следовательно, при этой коме наряду с введением инсулина необходимо и введение жидкостей.
3. Гипогликемическая кома. Может развиться как при тип I, так и при тип II СД вследствие передозировки инсулина, или когда введение инсулина не сопровождается приемом пищи. Гипогликемическая кома может возникнуть при вторичном гипопитуитаризме (обусловленном ангиопатией сосудов гипофиза), при диабетическом нефросклерозе (продлевает время циркуляции инсулина в крови). Основополагающим звеном этой комы является недостаток глюкозы в крови.
Поздние (хронические) осложнения СД. При хронической гипергликемии глюкоза запускает ряд патогенетических путей, которые повреждают инсулин-независимые клетки, изменяют внеклеточный матрикс,
что приводит к развитию хронических осложнений СД. Кратко представляем эти патогенетические пути:
а) Неферментное гликирование, неферментное, необратимое связывание производных глюкозы – дикарбонилов с аминогруппами внутриклеточных и внеклеточных белков и изменения свойств этих белков с образованием конечных продуктов гликирования (Advanced glycation end products (AGEs)). Отметим, что уровень гликированного гемоглобина HbA1C часто проверяется у больных сахарным диабетом. Он показывает эффективность гликемического контроля в течении последних 100-120 дней. Его высокий уровень указывает на плохой контроль уровня глюкозы и высокий риск хронических осложнений.
Внеклеточный AGEs связывается со специфическими рецепторами RACE макрофагов, Т-лимфоцитов,
эндотелиоцитов и гладких миоцитов стенки сосудов, что приводит к следующим изменениям:
- активация синтеза цитокинов и факторов роста (TGF-β, VEGF)→утолщение базальных мембран, пролиферация гладких миоцитов, ангиогенез (например, сетчатки)→гипертензия, ретинопатия,
12
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
-гиперпродукция свободных радикалов в эндотелиальных клетках→эндотелиальная дисфункция→
вазоконстрикция и адгезия тромбоцитов→гипертензия, атеросклероз, тромботические осложнения.
Кроме того, AGEs создают ловушку в стенках сосудов для других белков, например, альбуминов (связывает их), что нарушает эластичность стенки сосуда, способствует развитию атеросклероза и диабетической нефропатии.
б) С протеинкиназный путь - высокий уровень внутриклеточной глюкозы стимулирует de novo синтез диацилглицерола, что приводит к активации протеинкиназы C, вследствие чего происходит активация продукции VEGF, TGF-β, PAI-1 (ингибитор фибринолиза) эндотелием → диабетическая микроангиопатия,
протромботические осложнения. Кроме того, этот путь в нейронах приводит к увеличению содержания внутриклеточного натрия и кальция с соответствующими последствиями (повреждение нейронов).
в) Полиоловый путь - в инсулин-независимых тканях (нервы, хрусталик глаза, почки, сосуды) увеличение со-
держания внутриклеточной глюкозы запускает полиоловый путь метаболизма: посредством фермента альдозо-
редуктазы из глюкозы образуется сорбитол, а затем фруктоза. Это NADPH-зависимый фермент и вследствие его перерасхода нарушается активность глутатион-редуктазы, что приводит к развитию оксидативного стресса и повреждению эндотелия, шванновских клеток → диабетическая периферическая нейропатия, ангиопатия,
нефропатия. Накопление сорбитола и фруктозы в хрусталике, приводят к развитию катаракты.
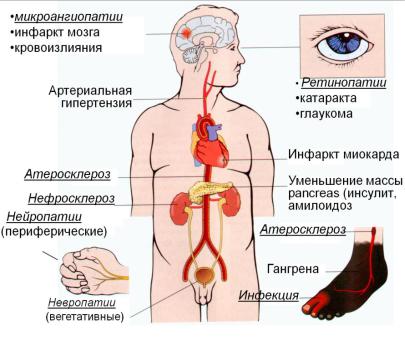
Поздние осложнения сахарного диабета
(рис. 12.10):
1. Макроангиопатии (атеросклеротическое поражение сосудов головного мозга, сердца,
почек, нижних конечностей, и других сосудов:
инсульты, инфаркты, «диабетическая стопа»).
В основе развития макроангиопатий лежит образование атеросклеротических бляшек в артериях большого и среднего диаметра. Они могут кальцифицироваться, изьязвляться, что способствует местному тромбообразованию и закупорке просвета сосудов. У больных СД преждевременно развивается атеросклероз, что связано также с накоплением холестерола, что является следствием:
а) гиперхолестеролемии,
б) уменьшения количества фосфолипидов в крови,
в) увеличения содержания ЛПНП и ЛПОНП в крови,
г) снижения липолитической активности сосудистой стенки,
д) снижения содержания липокаина в крови.
Уменьшение и/или закупорка просвета сосудов обусловлено также усилением синтеза тромбоксана, что способствует адгезии тромбоцитов и спазму сосудов.
Из-за высокого риска развития сосудистых осложнений при тип II СД Американская ассоциация кардиологов классифицировала его как сердечно-сосудистое заболевание.
2. Микроангиопатии (почки-диабетическая ангионефропатия, сетчатка - диабетическая ретинопатия, нервная ткань - диабетическая нейропатия). Длительная гипергликемия приводит к гликозилированию белков
13
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
организма. В результате клеточная деятельность нарушается и образуются аутоантитела к видоизмененным белкам с образованием иммунных комплексов.
Механизмы глюкотоксичности в клетках нервной системы, поджелудочной железы, почек, стенок сосудов,
в хрусталике глаза, в перицитах сетчатки, посредством запуска представленных сдвигов приводят к нарушениям микроциркуляции:
-накопление сорбита, маннита и др. в стенках сосудов,
-гликирование белков стенок сосудов,
-уменьшение образования NO,
-снижение активности Na+/K+ и Ca2+-зависимых АТФ-аз → ионный дисбаланс и гипергидратация клеток →
устойчивая вазоконстрикция,
-увеличение тромбогеного потенциала эндотелиоцитов,
-уменьшение соотношения простациклин/тромбаксан →микротромбозы.
3.Вторичный иммунодефицит,
4.гликирование гемоглобина и других белков,
5.нарушения зрения в результате поражения сетчатки (диабетическая ретинопатия) и хрусталика (катаракта).
14