

3 курс / Патологическая физиология / ОБЩАЯ_НОЗОЛОГИЯ_–_ОБЩЕЕ_УЧЕНИЕ_О_БОЛЕЗНИ,_ЭТИОЛОГИИ_И_ПАТОГЕНЕЗЕ
.pdfКлапаны |
Повышенная растяжимость |
Пролапс клапанов сердца, недостаточность кла- |
|
|
панного аппарата вен, варикоз, подострый бакте- |
|
|
риальный миокардит |
Сосуды |
Недостаточность эластиче- |
Расширение корня аорты, расслаивающие ане- |
|
ского компонента |
вризмы аорты, разрывы крупных сосудов, «хруп- |
|
|
кость» сосудов, легкость травматизации |
Пищеварите- |
Недостаточность эластиче- |
Хронические расстройства пищеварения |
льн. система |
ского компонента |
|
Мышцы |
Растяжимость |
Мышечная слабость; грыжи; растяжения, опуще- |
|
|
ния и выпадения внутренних органов |
Классификация наследственных болезней соединительной ткани основывается обычно на результатах работы McKusick, который проанализировал признаки, симптомы и морфологические изменения у большого числа больных. Однако классификация осложняется гетерогенностью этих синдромов. У больных, членов некоторых семей, отсутствует, например, один или несколько кардинальных признаков. В других семьях выявляют больных с двумя или тремя разными синдромами. Гетерогенность может быть обнаружена и среди членов одной семьи.
Синдром Элерса-Данлоса, или Элерса-Данло (СЭД) (синонимы: несовершен-
ный десмогенез, несовершенный десмогенез Русакова, синдром Черногубова-Элерса-Данлоса). Частичное клиническое описание этого синдрома впервые было сделано ещё в 1657 г. гол-
ландским хирургом Д. ван Мекреном с зарисовкой больного. Первая документальная фотография (больной позировал, сильно растянув кожу на груди) была сделана в 1880 г. В России синдром подробно описал А.Н. Черногубов (1891), назвав его генерализованным нарушением соединительной ткани. Однако он не продолжил этих наблюдений, а за границей был опубликован только реферат его работы. Позже этот синдром был описан Э. Элерсом (1901) и Х.А. Данло (1908) и назван по имени этих учёных.
CЭД представляет собой гетерогенную группу наследственных соединительнотканных заболеваний, общими клиническими признаками которых являются гипермобильность суставов, повышенная растяжимость кожи и хрупкость тканей. Эти изменения связаны с различными дефектами ряда коллагеновых белков, вследствие которых изменяются опорно-механические свойства соединительной ткани. Коллаген входит в состав практически всех органов и тканей, поэтому при СЭД клинически отмечается полисистемность поражения. Данные о частоте СЭД значительно варьируют – от 1:5000 до 1:560000. Тем не менее, некоторые авторы предполагают, что СЭД, возможно, является самым распространенным наследственным соединительнотканным заболеванием.
В разные периоды изучения СЭД существовали классификации, согласно которым выделяли от 3 до 11 типов синдрома. По мере накопления молекулярно-генетических данных возникла необходимость и возможность пересмотреть прежние варианты подразделения СЭД, и в 1997 году была предложена классификация, включающая 6 типов синдрома. Данная классификация на сегодняшний день максимально приближена к этиологическим факторам. По современным представлениям, все типы СЭД относятся к коллагенопатиям. При СЭД установлено вовлечение, по крайней мере, трёх типов: I, III и V.
Общая клиническая характеристика
Основными клиническими проявлениями при СЭД являются различные изменения со стороны кожи и суставов. Отмечается гиперрастяжимость кожи (рис. 6, 7).
Рис. 6. Гиперрастяжимость кожи |
Рис. 7. Гипермобильность суставов |
361
Заживление ран, даже незначительных порезов, протекает длительно. Особенно это касается послеоперационных швов, возможно их расхождение. В связи с расхождением краёв раны происходит формирование атрофичных рубцов (по типу «папиросной бумаги»). Обычно рубцы манифестируют, когда ребёнок начинает ползать или ходить, с локализацией на местах, подверженных механическому воздействию – колени, локти, лоб. Кожа при СЭД обычно тонкая.
В одной трети случаев выявляются маленькие плотные подкожные кистоподобные узелки, которые на рентгенограмме проявляются как кальцифицированные. Их можно прощупать на передневнутренней поверхности голеней и предплечий. Возможно, их возникновение связано с фиброзированием подкожных жировых долек вследствие нарушения кровообращения и последующей кальцификацией.
Дефект сосудистой стенки является причиной образования экхимозов, гематом и кровотечений. На местах повторных подкожных кровоизлияний могут формироваться особые припухлости – «подкожные псевдоопухоли», с возникновением характерной коричневой пигментации. Чаще всего они появляются на локтях и коленях. Геморрагические изменения возникают при нормальном коагуляционном статусе, что важно при дифференциальной диагностике. Данный аспект заболевания должен учитываться при оперативных вмешательствах.
Другим кардинальным симптомом СЭД является гипермобильность суставов (рис. 7). Она определяется на основании оценки подвижности суставов по шкале Бейгтона (Beighton et al., 1983). Шкала включает в себя 5 признаков – 4 парных и 1 непарный:
пассивное тыльное сгибание мизинца более 90° (по 1 баллу для каждой руки);
пассивное приведение большого пальца к сгибательной поверхности предплечья (по 1 баллу для каждой руки):
переразгибание локтевых суставов более 10° (по 1 баллу для каждой руки);
переразгибание коленей более 10° (по 1 баллу для каждой ноги);
наклон вперед, не сгибая коленей, с касанием пола ладонями (1 балл).
Гипермобильностью считается наличие 5 баллов и более по данной шкале.
Кроме этого характерны вывихи и подвывихи суставов, в том числе врожденный вывих бедра. Отмечаются хронические артралгии (без воспалительных изменений суставов). Отмечаются сколиоз, кифоз и их сочетание.
При СЭД в патологический процесс нередко вовлекаются другие органы и системы. Например, наблюдаются изменения со стороны глаз: миопия, кератоконус, голубые склеры, отслойка сетчатки. Особое значение имеют изменения со стороны сердца и крупных артерий. Типичным признаком является пролапс митрального клапана. К другим, более редким изменениям, относятся пролапсы других клапанов сердца и проксимальная дилятация аорты.
Мышечная гипотония, проявляющаяся задержкой моторного развития и быстрой утомляемостью, также является симптомом СЭД. Типичны грыжи, опущение внутренних органов (в частности, пролапс органов малого таза у женщин и выпадение прямой кишки у маленьких детей). К акушерским проблемам СЭД относятся преждевременный разрыв плодных оболочек, приводящий к преждевременным родам и послеродовые кровотечения.
Клинические варианты СЭД согласно последней классификации и критерии их диагностики
1. Классический тип (Classical). Наследуется по аутосомно-доминантному типу.
Основные диагностические критерии:
гиперрастяжимость кожи,
характерные атрофичные рубцы,
гипермобильность суставов.
Дополнительные диагностические критерии:
гладкость,
бархатистость кожи, подкожные псевдоопухоли,
подкожные узелки,
362
осложнения гипермобильности суставов (растяжения, вывихи и подвывихи, плоскостопие),
мышечная гипотония и задержка моторного развития,
лёгкое возникновение экхимозов,
проявления повышенной растяжимости и хрупкости тканей (грыжи, цервикальная недостаточность, опущение внутренних органов и др.),
хирургические осложнения (послеоперационные грыжи, расхождение швов, кровотечения во время и после операций).
Кроме того, диагностическую ценность имеет наличие родственников с аналогичными клиническими проявлениями.
Изменения со стороны кожи варьируют по степени тяжести (тяжёлые, средней тяжести, умеренные). Повторные подвывихи наиболее характерны для плечевых, височно-нижне-челюстных суставов и надколенников. Частой жалобой является повышенная утомляемость.
Несмотря на то, что классический тип составляет около 90% среди случаев СЭД всех типов, этиология этого заболевания до сих пор окончательно не установлена. За последние несколько лет появились сообщения об обнаружении мутаций в генах коллагена V типа, играющего важную роль в фибриллогенезе и регуляции диаметра коллагена I типа. Помимо мутаций в генах коллагена, причиной заболевания могут быть мутации в генах белков экстраклеточного матрикса, среди которых – декорин, люмикан, тенасцин Х. Таким образом, на сегодняшний день можно констатировать генетическую гетерогенность классического типа СЭД.
2. Гипермобильный тип СЭД (Hypermobility). Наследуется по аутосомно-доми- нантному типу.
Основные диагностические критерии:
проявления со стороны кожи (гиперрастяжимость и/или гладкая, бархатистая кожа),
генерализованная гипермобильность суставов.
Дополнительные диагностические критерии:
повторные вывихи суставов,
хронические боли в суставах либо конечностях;
выявление семейных случаев.
Всоответствии с прежней классификацией, этот подтип назывался «доброкачествен-ный гипермобильный тип», что подчеркивает доминирование выраженной гипермобильности суставов при отсутствии скелетных и относительно незначительных кожных проявлениях. Кожная растяжимость вариабельна, атрофичные рубцы не характерны.
Боли в суставах и мышцах начинаются рано, носят хронический и изнуряющий характер. Молекулярный дефект, лежащий в основе этого типа, на сегодняшний день окончательно неизвестен. Можно предполагать вовлечение коллагена III и I типов коллагена, однако не-обходимы дальнейшие исследования.
3. Сосудистый или васкулярный тип (Vascular). Наследуется по аутосомно-
доминантному типу.
Основные диагностические критерии:
тонкая, прозрачная кожа,
разрывы стенки артерий, кишечника и/или матки,
обширные кровоизлияния,
характерный фенотип.
Дополнительные диагностические критерии:
гипермобильность мелких суставов,
разрыв сухожилий и мышц,
косолапость,
варикозное расширение вен с ранней манифестацией,
артериовенозные каротидно-кавернозные фистулы,
363
пневмоторакс/пневмогидроторакс,
атрофия десневого края;
положительный семейный анамнез,
случаи внезапной смерти у близких родственников (многие пациенты не доживают до 50 лет из-за разрыва артерий или, что реже кишечника).
Наличие двух и более главных критериев с большой вероятностью указывает на диагноз васкулярного типа СЭД и является показанием для лабораторной диагностики.
Нередко отмечается характерный фенотип (узкий нос, тонкие губы, натянутая кожа, впавшие щеки и экзофтальм, обусловленный, в основном, снижением подкожно-жирового слоя). Надо отметить, что такой фенотип характерен для взрослых больных, а у детей он практически не выражен. Гипермобильность суставов обычно ограничена суставами пальцев.
Максимальная частота спонтанных артериальных разрывов приходится на третью-четвертую декады жизни, но они могут возникать и раньше. Чаще всего вовлечены артерии среднего калибра.
Беременность и роды могут осложняться разрывом матки и маточными кровотечениями, а также разрывом влагалища и промежности.
Именно артериальные разрывы являются наиболее частой причиной внезапной смерти. Поэтому в качестве диагностических мероприятий рекомендуются, по возможности, неинвазивные процедуры. При этом типе СЭД сравнительно хорошо изучены изменения на молекулярно-белковом уровне. Этиологическим фактором являются мутации в гене коллагена III типа.
4. Кифосколиотический тип (Kyphoscoliosis). Наследуется по аутосомнорецессивному типу.
Основные диагностические критерии:
генерализованная гипермобильность суставов,
выраженная мышечная гипотония при рождении,
прогрессирующий (с рождения) сколиоз, кифоз,
хрупкость склер,
разрывы глазного яблока.
Дополнительные диагностические критерии:
хрупкость тканей,
иногда атрофичные рубцы,
склонность к кровоизлияниям,
разрывы артерий,
марфаноидный фенотип,
микрокорнеа,
рентгенологически выявляемая выраженная остеопения,
положительный семейный анамнез ( в том числе больные сибсы).
Следует отметить, что наличие трёх основных критериев является основанием для предположения диагноза и требует лабораторного обследования. Причём мышечная гипотония может быть очень выраженной и обусловливает задержку моторного развития. Выраженный сколиоз приводит к тому, что во второй-третьей декаде жизни больные теряют способность передвигаться самостоятельно. Хрупкость тканей глаза является причиной разрыва глазного яблока при малейшей травме. Согласно последним данным, тяжёлые глазные осложнения встречаются гораздо реже, чем предполагалось ранее.
При этом типе СЭД дефект в синтезе коллагена заключается в изменении фермента лизилгидроксилазы, которая катализирует гидроксилирование боковых лизиновых цепей, не-обходимых для перекрестных связей между соседними коллагеновыми молекулами при формировании тройной спирали.
5. Артрохалазия (Arthrochalasis). Наследуется по аутосомно-доминантному типу.
364
Основные диагностические критерии:
тяжёлая генерализованная гипермобильность суставов с повторными вывихами (подвывихами),
врождённый вывих бедра.
Дополнительные критерии:
гиперастяжимость кожи,
хрупкость тканей,
атрофичные рубцы,
частые кровоизлияния,
мышечная гипотония,
кифосколиоз,
рентгенологически выявляемая умеренно выраженная остеопения,
врожденный вывих бедра (имеется у всех больных с биохимическим подтверждением диагноза),
низкий рост (является следствием выраженного кифосколиоза и/или вывиха бедра).
Причиной данного варианта СЭД являются мутации в генах α1 и α2 цепей коллагена I типа, которые возникают соответственно при А и В подтипах. В результате структурной мутации коллагенового гена становится невозможным функционирование N-терминальной пептидазы, что приводит к аномальному фибриллогенезу коллагена I типа.
6. Дерматоспараксис (Dermatosparaxis).Наследуется по аутосомно-рецессивному типу.
Основные диагностические критерии:
выраженная хрупкость кожи,
избыточная и отслаивающаяся кожа.
Дополнительные диагностические критерии:
мягкая, тестообразная консистенция кожи,
лёгкое образование кровоизлияний,
преждевременный разрыв плодных оболочек,
грыжи (пупочные, паховые), как правило крупных размеров.
Хрупкость кожи и образование кровоизлияний ярко выражены, в то время как заживление ран происходит обычно, без атрофичных рубцов. Избыток кожи на лице приводит к фенотипу, напоми-
нающим cutis laxa («вялая кожа»).
Название этого подтипа было взято по аналогии с заболеванием у крупного рогатого скота, овец и других животных с аналогичным изменением кожных покровов. К настоящему времени описано небольшое количество пациентов, и клинический спектр, видимо, может расширяться. Заболевание связано с мутациями в гене, кодирующем N-терминальную пептидазу проколлагена I, что приводит к дефициту данного фермента и нарушению фибриллогенеза.
Выяснение молекулярно-генетических причин и патогенеза различных типов СЭД является непростой, но интересной и важной задачей. Во-первых, СЭД представляет собой своеобразную модель для изучения сложных механизмов взаимодействия большого спектра молекул экстраклеточного матрикса в норме. Во-вторых, уточнение этиологических факторов СЭД позволит улучшить диагностику и прогноз течения заболевания.
Точная и своевременная молекулярно-генетическая диагностика СЭД является необходимым условием для принятия эффективных мер по профилактике на основе медико-генетического консультирования, пренатальной диагностики. Дальнейшие молекулярно-генетические исследования механизмов этиологии и патогенеза могут позволить перейти к разработке и применению генотерапевтических процедур, а следовательно, качественно новому этапу терапии больных СЭД-генотерапии.
365
Синдром Марфана
Синдром Марфана (англ. Marfan syndrome, болезнь Марфана) – аутосомно-доминантное генетическое заболевание, которое поражает соединительную ткань. Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Преимущественно эта болезнь наследуется по доминантному признаку и вызывается аномалией гена FBN1, кодирующего белок фибрилин-1. У каждого человека есть пара таких генов. Поскольку наследование происходит по доминантному типу, то люди, что наследуют один аномальный ген FBN1 от кого либо из родителей, будут поражены указанным заболеванием.
Синдром Марфана может появляться в умеренной и тяжёлой форме. Люди с этим заболеванием, как правило, высокие, с длинными конечностями и длинными худыми пальцами. Наиболее серьёзными осложнениями болезни является повреждение клапанов сердца и нарушение структуры стенок аорты. Также заболевание может влиять на лёгкие, глаза, твердую оболочку спинного мозга, скелет и твёрдое нёбо.
Кроме функций связующего белка, который служит опорой для ткани за пределами клетки, белок фибрилин связывается с другим белком, вследствие чего образуется трансформирующий фактор роста бета (TGF-β). TGF-β имеет негативное влияние на сосудистый тонус гладких мышц и нарушает развитие целостного внеклеточного матрикса.
Еще одной причиной развития болезни вследствие мутации гена ответственного за синтез фибрилина, сегодня учёные называют накопление избыточного количества TGF-β в лёгких, клапанах сердца и в аорте, что ослабляет ткани и вызывает симптомы болезни Марфана.
Болезнь получила название от имени Антуана Марфана, французского педиатра, который впервые описал симптомы заболевания в 1896 году, заметив черты синдрома у пятилетней девочки. Ген, который вызывает болезнь, был впервые обнаружен Франческо Рамиресом в 1991 г.
Симптомы
Хотя нет никаких уникальных симптомов болезни Марфана, однако, сочетание таких признаков как длинные конечности, дислокация хрусталика, аневризма корня аорты, вполне достаточно для того, чтобы с уверенностью поставить диагноз.
Поражение костной системы. Большинство видимых признаков болезни Марфана, связанные с костной системой. У многих людей с синдромом Марфана рост значительно выше среднего. Некоторые из них имеют длинные конечности с длинными тонкими пальцами рук и ног (арахнодактилия, рис. 8).
Кроме диспропорций развития конечностей и чрезмерного роста болезнь Марфана вызывает и другие нарушения в функционировании костной системы:
искривление позвоночника (сколиоз),
воронкообразная (внутрь) и килевидная (наружу) деформация грудной клетки (рис. 9),
чрезмерная гибкость суставов,
высокое нёбо,
неправильный прикус,
плоскостопие,
молоткообразная деформация пальцев стопы (когда суставы пальцев на ногах согнуты и напоминают, поэтому молоток,
сутулость,
появление беспричинных растяжек на коже (стрии),
у некоторых пациентов может появляться боль в суставах, костях и мышцах,
иногда возникают расстройства или нарушения речи (из-за высокого нёба и малых размеров челюсти),
вероятность развития остеоартрита в раннем возрасте.
366

Рис. 8. Арахнодактилия |
Рис. 9. Килевидная деформация груд- |
|
ной клетки |
Нарушение функций глаз и зрения. Болезнь Марфана может влиять на зрение и глаза. Обычно у пациентов наблюдаются астигматизм и близорукость, однако иногда фиксируется и дальнозоркость. Нарушение положения хрусталика в одном или обоих глазах (эктопия хрусталика, рис. 10), наблюдается у 80% больных.
Выявить указанные проблемы со здоровьем может офтальмолог с помощью щелевой лампы (метод биомикроскопии). При синдроме Марфана дислокация преимущественно имеет суперотемпоральний характер (вверх и наружу). Иногда проблемы со зрением возникают только после ослабления соединительной ткани, которое вызвано расслоением сетчатки. Еще одной из офтальмологических проблем, связанных с синдромом Марфана можно назвать раннюю (в молодом возрасте) глаукому.
Рис. 10. Эктопия хрусталика
Система кровообращения. Наиболее серьезными признаками и симптомами болезни Марфана, является нарушение деятельности сердечно-сосудистой системы. Чрезмерная усталость, одышка, нарушения ритма сердца, тахикардия (учащенное сердцебиение), стенокардия (которая сопровождается возникновением болевых ощущений в спине, плече или руке) – это те нарушения, которые наблюдаются при синдроме Марфана. Часто у пациентов, из-за нарушения кровообращения конечности (руки, ноги) холодные. Причинами для дальнейших обследований такого пациента может быть присутствие шумов в сердце, изменения на ЭКГ или присутствие симптомов стенокардии. Одной из причин регургитации (движения крови в противоположную сторону нормального направления), которая возникает из-за пролапса аортального или митрального клапанов сердца, можно назвать кистозную медиальную дегенерацию клапанов.
Однако основным признаком для дальнейшего детального исследования и изучения заболевания является расширенная аорта или аневризма аорты (выпячивание стенки аорты, рис. 11). Однако, иногда, очевидных проблем с сердечно-сосудистой системой не наблюдается, но ослабление соединительной ткани (через кистозную дегенерацию средней стенки сосудов – медии) вызывает аневризму или расслоения восходящей части аорты, что требует хирургического лечения. Расслоение аорты часто сопровождается болями в спине или груди и приводит к возникновению ощущения надрыва. Через нарушение функциональности соединительной ткани (что является патогенетическим механизмом развития синдрома Марфана) увеличивается частота случаев, при которых искусственный митральный клапан не приживается в организме больного. Именно поэтому необходимо проявлять чрезвычайную осторожность при лечении клапанов сердца. Предпочтение следует отдавать таким
367

мерам, которые направлены на восстановление функциональности больного клапана, а не на немедленную его замену.
Рис. 11. Аневризма аорты.
Во время беременности у женщин с синдромом Марфана, даже при отсутствии видимых отклонений в работе сердечно-сосудистой системы, высокий риск расслоения аорты, что может привести к летальному исходу, даже при своевременном лечении. Именно поэтому, если у женщины присутствует это заболевание, то перед зачатием необходимо пройти тщательное исследование и получить консультацию врача. А во время самой беременности каждые шесть-десять недель нужно проводить ЭХОКГ для определения диаметра корня аорты. Во многих случаях возможны естественные роды без осложнений, однако, лишь после исчерпывающего медицинского обследования и оценки всех возможных рисков.
Влияние на лёгкие. Болезнь Марфана является одним из факторов риска для спонтанного возникновения пневмоторакса. Больной пневмотораксом испытывает резкую боль в груди, дышит часто и поверхностно, наблюдается выраженная отдышка. Часто проявляется бледность или синюшность кожного покрова, в частности лица (цианоз). Если заболевание не лечить, оно может привести к смерти больного.
Кроме этого, синдром Марфана может быть связан с такими заболеваниями лёгких как апноэ во сне (это прекращение вентиляции лёгких во время сна, более чем на 10 секунд) и другими идиопатическими (с неустановленной причиной) обструктивными болезнями лёгких.
Влияние на ЦНС. Одним из последствий болезни Марфана, который может негатив-но повлиять на качество жизни человека (хотя он не представляет угрозы жизни) является дуральная эктазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка – мембраны, которая окутывает спинной мозг.
В течение длительного времени симптомы дуальной эктазии (боль в пояснице, в ногах, в области живота и таза, другие неврологические симптомы в нижних конечностях или головная боль) могут не проявляться. Или же резко исчезают, когда человек лежит на плоской ровной поверхности, на спине. При болях такого типа врачи обычно назначают рентгенографию поясничного отдела позвоночника, хотя, как правило, дуральную эктазию невозможно заметить с помощью рентгенологического исследования на ранних стадиях. Именно поэтому, ухудшение симптомов и отсутствие, какой либо другой причины боли создает необходимость проведения исследования с помощью магнит- но-резонансной томографии (МРТ) поясничного и крестцового отделов позвоночника.
Другие неврологические проблемы, связанные с синдромом Марфана – это дегенеративные заболевания межпозвоночных дисков и костей спины.
Также синдром Марфана является важным фактором вызывающим развитие дисфунк-ции
ВНС.
Лечение. На сегодня лекарства от болезни Марфана не существует, однако за последние десятилетия продолжительность жизни с этим заболеванием значительно возросла. Особенно важным является профилактика заболевания, даже для маленьких детей, которая дол-жна быть направлена на замедление развития аневризмы аорты.
Физиотерапевт может также помочь улучшить функциональность опорно-двигатель-ной системы и уменьшить травматизм у лиц с болезнью Марфана.
368
Несовершенный остеогенез
Несовершенный остеогенез (НО) (лат. osteogenesis imperfecta; синонимы: «несовершенное костеобразование», болезнь «хрустального человека», «врожденная недостаточность и ломкость кости», болезнь Лобштейна-Вролика) – врожденное метаболическое заболевание кости. НО встречается с частотой 1 на 10000-20000 живых новорождённых. Заболевание характеризуется вовлечением в патологический процесс костей и мышц, вызывается дефектом в синтезе коллагена I типа, который является главным компонентом матрикса кости.
Патогенез
Выявлено более 200 мутаций в генах COL1A1 или COL1A2, кодирующих составные цепи коллагена I типа. Наиболее лёгкие и распространенные формы НО обусловлены преждевременной остановкой кодонов или дефектом, возникающим при сплайсинге РНК, что ведёт к нарушению остеобластами биосинтеза I типа коллагена. В результате биосинтеза дефектного коллагена у пациентов с несовершенным остеогенезом нарушается формирование костного матрикса, при этом изменяется баланс между резорбцией и формированием кости в сторону преобладания резорбции.
Известно, что в норме, во время роста ребёнка, преобладает формирование костной ткани, увеличиваются объём и прочность костных трабекул. При несовершенном остеогенезе более тонкие первичные трабекулы разрушаются, частично за счёт неустойчивости коллагена и, возможно, из-за более высокого, чем в норме, физического напряжения внутри кости, которое стимулирует остеоциты к активизации локального ремоделирования, что делает эти участки уязвимыми для микротрещин. Все эти механизмы могут способствовать нарушению первичной трабекулярной структуры кости и, таким образом, делать её еще более слабой. Поэтому у растущего ребенка с несовершенным остеогенезом – тонкая кость с нарушенной трабекулярной структурой, тонким корковым слоем и с высоким уровнем ремоделирования. В результате этого возникают деформации, а также увеличивается риск переломов.
Клиническая картина
Несовершенный остеогенез зависит от тяжести заболевания и может проявляться летальными вариантами, явными аномалиями скелета у детей или иметь типичную манифестацию у людей зрелого возраста.
Из-за различной и многообразной клинической картины несовершенный остеогенез часто не диагностируют, пропускают или поздно верифицируют, или же пациента лечат под другим диагнозом.
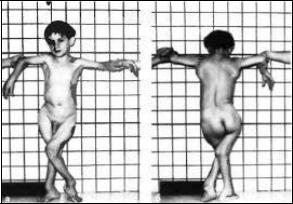
У детей с НО выявляют задержку физического развития, сколиоз, прогрессирующие деформации длинных костей, тугоухость, патологию прорезывания зубов. Тяжесть заболевания обусловливается частотой переломов, прогрессирующей деформацией, хронической болью в костях и потерей подвижности (рис. 12).
Рис. 12. Внешний вид больного с несовершенным остеогенезом.
Классификация несовершенного остеогенеза
В настоящее время широко применяют классификацию D. Sillence, дополненную M. Ramachandran, которая базируется на данных клинического и рентгенологического обследования пациента и позволяет выделить 4 генетических типа заболевания. Следует отметить, что во время формулирования классификации считали, что несовершенный остеогенез наследуется как по ауто-
369
сомно-доминантному, так и аутосомно-рецессивному типам, однако сейчас доказано, что преобладает аутосомно-доминантный тип наследования с семейным мозаицизмом. Очень редко наблюдается несовершенный остеогенез с аутосомно-рецессивным типом наследования. В классификации, представленной M. Ramachandran, также учитывается состояние дентиногенеза.
На сегодня выделено ещё четыре типа несовершенного остеогенеза (V, VI, VII, VIII), которые не связаны с патологией коллагена I типа и пока не внесены в Международную классификацию остеохондропатий.
Характеристика типов несовершенного остеогенеза
І-й тип. Коллаген нормального качества, но вырабатывается в недостаточных количествах.
Кости легко ломаются, в особенности до пубертата.
Лёгкое искривление спины.
Слабость связочного аппарата суставов.
Пониженный мышечный тонус.
Обесцвечивание склер, обычно придающие им голубовато-карий цвет.
Ранняя потеря слуха у некоторых детей.
Слегка выступающие глаза.
Также различают 1-й тип A и 1-й тип В по наличию или отсутствию несовершенного дентиногенеза (характеризуемый опаловыми зубами; отсутствует в IA, присутствует в IB). Помимо повышенного риска фатальных переломов костей, ожидаемая продолжительность жизни в пределах нормы.
II тип (перинатально-летальный) – это самый тяжёлый вариант болезни. Коллаген не-
достаточного количества или качества.
Большинство больных умирает на протяжении первого года жизни по причине дыхательной недостаточности или внутричерепного кровоизлияния.
Трудности с дыханием в связи с недоразвитыми лёгкими.
Тяжёлые деформации кости и невысокий рост.
2-й тип может быть далее разбит на подклассы A, B, C, различаемые радиографическим анализом длинной трубчатой кости и рёбер.
III тип. Коллаген в достаточных количествах, но недостаточного качества.
Кости ломаются легко, иногда даже при рождении.
Деформация костей, часто тяжёлые.
Возможны проблемы с дыханием.
Невысокий рост, искривление позвоночника, иногда бочковидная грудная клетка.
Слабость связочного аппарата суставов.
Слабый мускульный тонус в руках и ногах.
Обесцвечивание склер.
Иногда ранняя потеря волос.
3-й тип выделяется из других классификаций, будучи типом «прогрессивной деформации»,
где новорождённый имеет лёгкие симптомы при рождении и развивает вышеуказанные симптомы в процессе жизни. Продолжительность жизни может быть нормальной, хотя и с тяжёлыми физическими препятствиями. Как правило, дети с III типом несовершенного остеогенеза не способны к самообслуживанию и заканчивают жизнь в инвалидном кресле.
IV тип. Коллаген достаточного количества, но недостаточно высокого качества.
Кости ломаются легко, особенно до пубертата.
Невысокий рост, искривления позвоночника и бочковидная грудная клетка.
Деформация костей в диапазоне от слабой до средней.
Ранняя потеря волос.
Подобно 1-му типу, 4-й тип может быть далее разделён на подклассы IVA и IVB, которым характерно отсутствие (IVA) или наличие (IVB) несовершенного дентиногенеза.
370