

6 курс / Неонатология / Детские болезни Баранов А.А. 2009
.pdfНеонатальный скрининг недостаточности 21-гидроксилазы
Диагностика недостаточности 21-гидроксилазы основывается на клинических признаках заболевания. В первые недели жизни ребѐнка заподозрить заболевание позволяет наличие вирилизации наружных половых органов у девочек и развитие синдрома потери соли у детей обоего пола. Вирильную форму заболевания у мальчиков диагностируют только к 4-5 годам жизни при развитии симптомов преждевременного полового развития. Костный возраст при этом уже значительно опережает хронологический. Подобный клинический подход к диагностике недостаточности 21-гидроксилазы приводит к большому количеству ошибок. До 30% девочек с тяжѐлыми признаками вирилизации наружных половых органов ошибочно регистрируют как имеющих мужской пол. До 35% мальчиков с сольтеряющей формой заболевания погибают в первые недели жизни, так как заболевание вовремя не распознают. Избежать диагностических ошибок позволяет неонатальный скрининг на выявление недостаточности 21-гидрокси- лазы. В основе скрининга лежит определение содержания 17а-гид- роксипрогестерона в сухом пятне крови на фильтровальной бумаге. К 1991 г. программу неонатального скрининга внедрили в 29 странах мира. Проведение этой программы позволило определять реальную частоту недостаточности 21гидроксилазы и еѐ форм в различных популяциях.
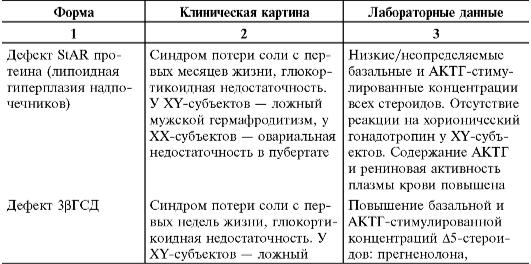
Различные формы врождѐнной дисфункции коры надпочечников представлены в табл. 18-5.
Таблица 18-5. Дифференциальная диагностика различных форм врождѐнной дисфункции коры надпочечников

Синдром потери соли часто приходится дифференцировать с пилоростенозом, пилороспазмом и острыми заболеваниями, сопровождающимися рвотой. Неправильное строение наружных половых органов требует исключения истинного и ложного гермафродитизма. Синдром внутриутробной вирилизации у девочек иногда может быть обусловлен приѐмом некоторых препаратов женщинами во время беременности. Гормональная и молекулярно-генетическая диагностика позволяет диагностировать и уточнять форму адреногенитального синдрома.
Лечение
Лечение врождѐнных нарушений надпочечникового стероидогенеза
Общие цели лечения всех форм врождѐнных нарушений надпочечникового стероидогенеза:
•заместить дефицит стероидов, секреция которых снижена в результате ферментативного дефекта;
•снизить концентрацию стероидов, секреция которых повышена в результате ферментативного дефекта;
•подавить избыточную секрецию АКТГ, используя минимально эф-
фективные дозы глюкокортикоидов;
•оптимизировать рост больных;
•предотвратить вирилизацию наружных половых органов;
•обеспечить нормальное половое созревание и фертильность.
Терапия недостаточности 21-гидроксилазы
Основным методом терапии недостаточности 21-гидроксилазы считают применение глюкокортикоидов, подавляющих гиперсекрецию АКТГ и нормализующих выработку андрогенов надпочечниками. Используются различные медикаментозные препараты, обладающие глюкокортикоидной активностью: преднизолон, кортизон, дексаметазон. Пролонгированные синтетические глюкортикоидные препараты (преднизолон, дексаметазон) оказывают негативное влияние на процессы роста. Их пролонгированный эффект может быстро привести к симптомам передозировки. Для детей с открытыми зонами роста, особенно младшего возраста, наиболее оптимальными препаратами следует считать таблетированные формы гидрокортизона (например, «Кортеф»). Первоначальная суточная доза гидрокортизона, необходимая для подавления АКТГ у детей первого года жизни, может достигать 20 мг/м2. Однако следует избегать длительного применения этих доз у ребѐнка. Применение гидрокортизона в дозе 25 мг/м2 в течение года приводит к выраженной задержке или остановке роста! В случае длительной передозировки глюкокортикоидов в младшем возрасте отставание в росте ликвидировать невозможно, даже на фоне снижения дозы препаратов. В среднем суточная доза гидрокортизона должна составлять 1015 мг/м2. Препарат назначают 3 раза в день в равных дозах (в 7.00, 15.00, 22.00). Всем детям с сольтеряющей формой 21-гидроксилазной недостаточности необходимо дополнительное назначение минералокортикоидных препаратов.
К показаниям для назначения минералокортикоидной терапии относят:
•развитие клинических симптомов сольтеряющего криза;
•высокая концентрация калия в сыворотке крови при отсутствии
клинических симптомов потери соли;
• высокая рениновая активность плазмы крови при нормальной кон-
центрации калия и отсутствии клинических симптомов потери соли.
Доза флудрокортизона составляет 0,05-0,3 мг/сут. Такая терапия позволяет компенсировать недостаток минералокортикоидов, добиться более быстрого подавления избыточной секреции АКТГ при использовании менее высоких доз глюкокортикоидов. При наличии минералокортикоидной недостаточности потребность в минералокортикоидах максимальна у детей первого года жизни и составляет 0,1-0,3 мг/сут. Суточную дозу назначают в три приѐма (в 7.00, 15.00, 23.00). Дополнительно вводят в рацион избыточное количество поваренной соли - до 2 г/сут.
В дальнейшем необходимость продолжения терапии минералокортикоидами основывается на показателях рениновой активности плазмы крови. У детей старшего возраста дозу минералокортикоидов снижают до 0,05-0,15 мг/сут. Суточную дозу назначают в два приѐма (в 8.00 и 18.00).
Контроль адекватности терапии
Контроль адекватности терапии недостаточности 21-гидроксилазы основан на показателях физического развития и данных гормонального исследования. Детей первых 2 лет жизни должен осматривать педиатр-эндокринолог каждые 3 мес. Детей старше 2 лет необходимо обследовать каждые 6-12 мес. При осмотре тщательно фиксируют показатели массы тела и роста. Определяют концентрации 17b-гидроксипрогестерона и электролитов. Проводят исследование рениновой активности плазмы крови. У детей старшего возраста ежегодно исследуют костный возраст, определяют содержание тестостерона.
Адекватная терапия глюкортикоидами обеспечивает нормальные темпы роста и костного созревания.
Терапия при ургентных состояниях
Родители пациентов с недостаточностью 21-гидроксилазы должны быть информированы об изменениях лекарственной терапии в неотложных и стрессовых ситуациях. Эту информацию необходимо выдать в виде письменных рекомендаций дополнительно к официальным медицинским выпискам. Дети старшего возраста и подростки должны
иметь при себе карту, в которую необходимо внести рекомендации о терапии в условиях неотложных ситуаций.
При интеркуррентных заболеваниях всем пациентам с высокой (>38,5 ?С) температурой тела, рвоте, травмах, хирургических вмешательствах необходимо дополнительное введение гидрокортизона. При стрессорных физических нагрузках (участие в соревнованиях) может возникнуть потребность в увеличении дозы глюкокортикоидов. Эмоциональные и
психические стрессы (экзамены) не требуют дополнительного введения препаратов.
В период стресса дозу глюкортикоидов необходимо увеличить в 2-3 раза. В тех случаях, когда пероральный приѐм препаратов не возможен, следует обеспечить парентеральное введение гидрокортизона.
При тяжѐлых интеркуррентных заболеваниях и оперативных вмешательствах следует применять внутримышечное введение гидрокортизона в дозе 3-5 мг/кг на инъекцию каждые 4-6 ч и дополнительное внутривенное капельное раствора натрия хлорида в количестве 150 мл/кг в сутки. При этом 25% указанного объѐма жидкости необходимо ввести в первые 2 часа терапии.
Хирургическая стратегия коррекции наружных половых органов у девочек
Хирургическая коррекция наружных половых органов производят девочкам с симптомами внутриутробной вирилизации. Цель хирургической коррекции - добиться соответствия наружных половых органов избранному (женскому) полу, ликвидировать возможные анатомические препятствия, затрудняющие отток мочи, создать условия для нормальной репродуктивной функции.
Пренатальная диагностика и лечение недостаточности 21-гидрокси- лазы
Недостаточность 21-гидроксилазы в классическом и неклассическом варианте считают одним из самых распространѐнных заболеваний с аутосомнорецессивным типом наследования. Разработка метода исследования содержания 17a-гидроксипрогестерона в капле крови, нанесѐнной на фильтровальную бумагу, позволила проводить массовый скрининг новорождѐнных в различных популяциях. В результате этих масштабных исследований было установлено, что средняя заболеваемость в мире составляет 1:13 500. В то же время в некоторых популяциях эта частота намного выше.
Профилактика
Как и при всех аутосомно-рецессивных наследственных заболеваниях, уменьшение частоты родственных браков сопровождается снижением заболеваемости.
Прогноз
Скрининг новорождѐнных и адекватная терапия с первых дней жизни значительно улучшают прогноз заболевания и социальную адаптацию детей. Пренатальная диагностика и лечение позволяют к моменту рождения избежать вирилизации наружных половых органов у девочек с недостаточностью 21-гидроксилазы.
Сахарный диабет
СД - группа заболеваний обмена веществ различной этиологии, характеризующихся хронической гипергликемией, возникающей в результате нарушения секреции или действия инсулина, либо обоих факторов одновременно (ВОЗ, 1999).
Исследовательская группа ВОЗ с учѐтом новых данных пересмотрела классификацию СД. Эта классификация включала клинические формы заболевания и классы статистического риска.
ЭТИОЛОГИЧЕСКАЯ КЛАССИФИКАЦИЯ САХАРНОГО ДИАБЕТА (ВОЗ, 1999)
• СД 1-го типа (деструкция β-клеток, приводящая обычно к абсолютной инсулиновой недостаточности):
-аутоиммунный;
-идиопатический.
•СД 2-го типа (с преимущественной инсулинорезистентностью и относительной инсулиновой недостаточностью или преимущественным дефектом секреции инсулина с инсулинорезистентностью или без неѐ).
•Гестационный СД.
•Другие специфические типы СД:
-генетические дефекты функции β-клеток;
-генетические дефекты в действии инсулина;
-болезни экзокринной части поджелудочной железы;
-эндокринопатии;
-СД, индуцированный лекарствами или другими химическими веществами;
-СД, индуцированный инфекциями;
-необычные формы иммуноопосредованного СД;
-другие генетические синдромы, иногда сочетающиеся с СД.
СД 1-го и 2-го типов - наиболее распространѐнные формы СД. Они различаются по клиническим, эпидемиологическим и иммунологическим характеристикам, характеру секреции инсулина, ассоциации с генетическими маркѐрами.
СД 1-го типа наиболее часто наблюдают у детей и лиц молодого возраста, хотя это заболевание может манифестировать в любом возрасте. Аутоиммунный СД характеризуется деструкцией β-клеток, наличием аутоантител, абсолютной инсулиновой недостаточностью, полной
инсулинозависимостью, тяжѐлым течением со склонностью к кетоацидозу, ассоциацией с генами главного комплекса гистосовместимости HLA. Случаи заболевания идиопатическим СД регистрируют обычно у лиц, не относящихся к европейской расе, с деструкцией β- клеток, склонностью к кетозу, но неизвестным патогенезом.
СД 2-го типа среди взрослых является доминирующим. В детском возрасте его наблюдают крайне редко. СД 2-го типа в детском возрасте чаще протекает бессимптомно или с минимальной клинической симптоматикой. В то же время при инфекционных заболеваниях или сильных стрессах иногда может развиться кетоацидоз. В развитии заболевания у детей основное значение придают генетическому фактору. Монозиготные близнецы в 100% случаев конкордантны (сходны) по СД 2-го типа. У родителей также в большинстве случаев диагностируют СД 2-го типа, особенно при исследовании теста на толерантность к глюкозе. Большое значение в инициации заболевания имеют привычки поведения, такие как переедание и сниженная физическая активность. Внутриутробная задержка развития с дефицитом массы тела, а также недостаточное питание в раннем постнатальном периоде также могут способствовать развитию СД 2-го типа в детском возрасте из-за избыточного кормления ребѐнка, приводящего к формированию ожирения, гиперинсулинизма, инсулинорезистентности.
Длительное время считали, что для детского возраста характерна лишь одна форма - СД 1-го типа. Однако исследования последнего десятилетия убедительно показали, что наряду с доминирующим СД 1-го типа, в детском возрасте диагностируют и более редкие сочетания СД с генетическими синдромами, СД 2-го типа, преобладающий у взрослых, а также MODY тип, считавшийся специфичным лишь для юношеского возраста. В настоящее время заболеваемость СД 1-го типа у детей в России составляет 9,24 на 100 000 детского населения.
ЭТИОЛОГИЯ
В основе развития СД 1-го типа лежит генетическая предрасположенность. Об этом свидетельствуют семейные случаи заболевания, а также высокая частота повторных случаев заболевания среди монозиготных близнецов.
Семейная концентрация СД 1-го типа (или частота повторных случаев заболевания в семьях больных) определяется следующими факторами:
•частотой СД в популяции;
•количеством больных и здоровых родственников, степенью их род-
ства;
• возрастом возникновения СД у пробанда, в некоторых случаях -
полом пробанда;
• возрастом обследуемых родственников, в некоторых случаях - их

полом.
В табл. 18-6 представлены эмпирически полученные показатели риска развития СД 1-го типа в различных группах родственников для популяций с высоким уровнем заболеваемости (0,4%).
Таблица 18-6. Эмпирический риск развития заболевания у родственников пациентов с сахарным диабетом 1-го типа (Эйзенбарт, 1994)
Как показали многочисленные исследования последних лет, СД 1-го типа - аутоиммунное заболевание у генетически предрасположенных лиц, при котором длительно текущий хронический лимфоцитарный инсулит приводит к деструкции β-клеток с последующим развитием инсулиновой недостаточности.
Для начала аутоиммунного процесса необходим инициирующий или провоцирующий фактор внешней среды (триггер). На современном этапе не существует единого и несомненного взгляда на природу такого фактора. В настоящее время выделяют наиболее вероятные факторы, принимающие участие в запуске процессов разрушения островковых клеток.
•Вирусы: Коксаки B, краснухи, паротита, энтеровирусы, ротавирусы, цитомегаловирус, Эпстайна-Барр, ECHO и др.
•Факторы питания: коровье молоко и смешанное вскармливание на основе коровьего молока, продолжительность грудного вскармливания, нитраты.
•Воздействие токсинов.
ПАТОГЕНЕЗ
Механизмы повреждения β-клеток вирусами:
• прямое разрушение (лизис) β-клеток в результате инфицирования
вирусом;
•механизм молекулярной мимикрии, при котором иммунный ответ, направленный на вирусный Аг, сходный с собственным Аг β-клетки, повреждает и саму островковую клетку;
•нарушение функции и метаболизма β-клетки, в результате чего на
еѐ поверхности экспрессируются аномальные Аг, что приводит к запуску аутоиммунной реакции;
• взаимодействие вируса с иммунной системой.
Стадии развития СД 1-го типа представлены на рис. 18-7.
Аутоантитела к различным структурам β-клеток рассматривают как иммунологические маркѐры β-клеточной деструкции.
Инсулин - главный регулирующий обмен веществ гормон, конечным результатом действия которого является обеспечение энергетических и пластических процессов в организме. К органам-мишеням действия инсулина относят печень, мышечную и жировую ткани. Инсулин может оказывать анаболическое и антикатаболическое действия. Анаболический эффект инсулина реализуется через стимуляцию синтеза гликогена и жирных кислот в печени, триглицеридов в жировой ткани, белка и гликогена в мышечной ткани. Антикатаболическое действие инсулина заключается в подавлении процессов гликогенолиза, глюконеогенеза (образование глюкозы из жиров и белков) и кетоногеза (образование кетоновых тел). В организме существуют и инсулиннезависимые ткани (почки, головной мозг, шванновские клетки периферических нервов, ткань хрусталика, артерии, сетчатка), в которых для переноса глюкозы внутрь клетки инсулин не требуется.
Механизм действия инсулина заключается в активации транспорта глюкозы через мембрану клетки, а также стимуляции различных ферментов, участвующих на разных стадиях в обменных процессах. Действие инсулина осуществляется при связывании со специфическими рецепторами на цитоплазматических мембранах.
Все клинические симптомы обусловлены недостатком выработки и действия инсулина. У детей это обусловлено преимущественно гибелью β-клеток поджелудочной железы, т.е. имеет место абсолютная недостаточность инсулина.
КЛИНИЧЕСКАЯ КАРТИНА
СД может развиться у ребѐнка в любом возрасте. В течение первых месяцев жизни заболевание наблюдают редко. Риск увеличивается после 9 мес, достоверно нарастая после 5 лет и в пубертатном возрасте, и несколько снижается у взрослых.