

6 курс / Неонатология / Детские болезни Баранов А.А. 2009
.pdfУчитывая высокую вероятность гипогликемии, в схему лечения включают введение 5-20% раствора глюкозы по возможности под контролем еѐ содержания в плазме крови.
В зависимости от этиологии острой надпочечниковой недостаточности проводят лечение основного заболевания (антибиотикотерапию и др.).
Летальность в результате адреналового криза составляет около 50% и приходится на первые сутки заболевания.
После стабилизации состояния следует продолжить парентеральное введение глюкокортикоидов и 0,9% раствора натрия хлорида в течение нескольких дней под контролем гемодинамических показателей и содержания электролитов. Затем постепенно снижают дозу глюкокортикоидов и переходят к пероральному приѐму препаратов, дополняя заместительной терапией минералокортикоидами (флудрокортизон) в заместительных дозах. Далее необходимо уточнить наличие хронической надпочечниковой недостаточности и перейти к постоянной адекватной заместительной терапии.
Профилактика
Основной задачей первичной профилактики аддисонического криза, а следовательно, смертности в группе пациентов с хронической надпочечниковой недостаточностью является своевременная адекватная терапия основного заболевания. Необходимо осуществлять регулярный медицинский контроль за адекватностью заместительной терапии. Огромную роль играет обучение пациентов и членов их семей самоконтролю, правилам поведения в различных ситуациях, потенциально провоцирующих развитие адреналового криза. При интеркуррентных заболеваниях, травмах, хирургических вмешательствах, стрессе дозу глюкокортикоидов следует увеличивать в 2 раза с последующим постепенным возвратом к прежней заместительной дозе. В мировой практике рекомендуют ношение специальных браслетов, на которых отражена информация о болезни пациента и основные пункты оказания скорой помощи: введение глюкокортикоидов, экстренная госпитализация для дальнейшей квалифицированной медицинской помощи.
ИЗОЛИРОВАННАЯ МИНЕРАЛОКОРТИКОИДНАЯ
НЕДОСТАТОЧНОСТЬ
Изолированный гипоальдостеронизм в отсутствии дефицита других гормонов коры надпочечников проявляется клинической картиной потери соли. При данной патологии, в отличие от других форм надпочечниковой недостаточности, система гипоталамус-гипофиз-надпочечники интактна. Снижение концентрации альдостерона приводит к повышению активности только ренин-ангиотензиновой системы.
Выделяют 3 группы изолированной минералокортикоидной недостаточности:
• Врождѐнный первичный гипоальдостеронизм.
•Приобретѐнный вторичный дефицит альдостерона.
•Псевдогипоальдостеронизм.
Врождѐнный первичный гипоальдостеронизм
Врождѐнный первичный гипоальдостеронизм - редкая аутосомнорецессивная патология, характеризующаяся синдромом потери соли и задержкой физического развития. Данное заболевание обусловлено нарушением биосинтеза альдостерона вследствие дефицита фермента альдостеронсинтазы. В отличие от дефицита других ферментов стероидогенеза, дефицит альдостеронсинтазы не приводит к гиперплазии надпочечников, так как синтез кортизола при данном синдроме не нарушен. На рис. 18-4 представлены два последних этапа биосинтеза альдостерона, протекающие в клубочковой зоне коры надпочечников.
Известно, что синтез кортикостеронметилоксидазы (КМО) 1 и 2 типов происходит с одного гена CYP11B2, расположенного на хромосоме 8 (8q21). В зависимости от конкретной мутации поражается либо 18-гид- роксилазная активность фермента, либо альдегидсинтазная активность. Следовательно, выделяют 2 формы дефицита альдостеронсинтазы: недостаточность КМО-1 и КМО-2. Эти формы заболевания отличаются друг
от друга только гормональным профилем. При дефиците КМО-1 определяют низкие концентрации как альдостерона, так и 18-гид- роксикортикостерона, тогда как при дефиците КМО-2 значительно повышено содержание 18гидроксикортикостерона, а концентрация альдостерона низкая. Дифференциально-диагностическим критерием этих состояний служит отношение 18-гидрокси- кортикостерона к альдостерону: при дефиците КМО-1 этот показатель меньше 10, а при дефиците КМО-1 - превышает 100.
Клинические признаки не зависят от формы заболевания.
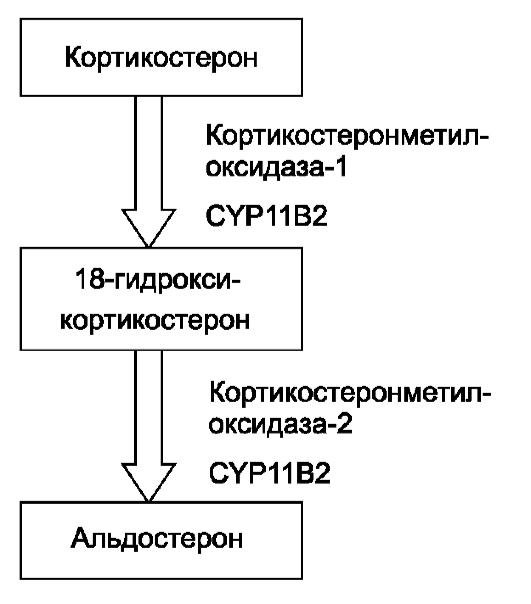
Рис. 18-4. Биосинтез альдостерона.
У новорождѐнных возникает рвота. Ребѐнок отказывается от пищи, перестает прибавлять в массе тела - развивается дегидратация. По данным лабораторных исследований, отмечают гиперкалиемию, иногда гипонатриемию, высокую активность ренина плазмы крови. У данных пациентов с возрастом наблюдают положительную динамику относительно синдрома потери соли, однако отмечают задержку физического развития. При лечении пациентов с дефицитом альдостеронсинтазы используют добавление к пище хлористого натрия (поваренной соли) и минералокортикоидные препараты (флудрокортизон в дозе 0,05-0,1 мг/сут).
Приобретѐнный вторичный дефицит альдостерона
Приобретѐнный вторичный дефицит альдостерона обусловлен снижением биосинтеза ренина в почках. В результате отсутствия стимуляции клубочковой зоны надпочечников ренин-ангиотензиновой системой снижается синтез альдостерона. Данное заболевание характеризуется хронической бессимптомной гиперкалиемией и некоторым снижением функций почек.
Однако у части пациентов могут развиваться мышечная слабость и сердечные аритмии. Вторичный гипорениновый гипоальдостеронизм наблюдают у пациентов с СД, при СКВ, миеломной болезни, почечном амилоидозе, циррозе печени, серповидноклеточной анемии, вегетативной полиневропатии и СПИДе.
Причиной заболевания является поражение юкстагломерулярного аппарата почек, что приводит к сниженной секреции ренина. Существует несколько теорий, объясняющих гипоренинемию. Например, длительно существующая гиперволемия приводит к необратимому подавлению функции юкстагломерулярного аппарата. Другой причиной гипоренинемии может быть недостаточная активность вегетативной нервной системы, например у пациентов с СД. Третьим возможным механизмом может стать нарушение преобразования проренина в ренин вследствие дефицита калликреина или ПгI2.
Для пациентов с гипорениновым гипоальдостеронизмом характерна особая форма почечного тубулярного ацидоза. В развитии ацидоза играет роль не только дефицит минералокортикоидов, но и гиперкалиемия, которые снижают почечный аммониогенез, уменьшают секреторную активность дистального отдела нефрона для ионов Н+.
Диагноз вторичного гипоренинемического гипоальдостеронизма необходимо рассматривать у всех пациентов с хронической гипокалиемией. Клинический диагноз подтверждают низкое содержание альдостерона в крови в сочетании с низкой плазменной активностью ренина. Результаты стимуляционных диагностических тестов, вызывающих активацию ренин-ангиотензин- альдостероновой системы (ортостатическая проба, проба с фуросемидом), отрицательные.
Терапия гипоренинового гипоальдостеронизма направлена на коррекцию гиперкалиемии. У пациентов с умеренной гиперкалиемией без изменений на ЭКГ необходимо проводить мониторирование электролитов и ЭКГ. Таким пациентам нужно рекомендовать диету с ограничением калия и предостерегать их от приѐма препаратов, вызывающих гиперкалиемию (β- адреноблокаторы, ингибиторы АПФ, гепарин натрия, калийсберегающие диуретики, ингибиторы циклооксигеназы). Терапию минералокортикоидами проводят пациентам с выраженной гиперкалиемией, без артериальной гипертензии и застойной сердечной недостаточности.
Псевдогипоальдостеронизм
Псевдогипоальдостеронизм - состояние, характеризующееся клинической картиной синдрома потери соли, но сопровождающееся высокими концентрациями альдостерона и ренина. Причиной данной патологии считают нарушение механизма действия альдостерона. Выделяют псевдогипоальдостеронизм с аутосомно-рецессивным типом наследования, причиной которого является патология амилоридчувствительных натриевых каналов в дистальных отделах нефрона, что приводит к повышенной экскреции натрия из организма. Выявлены мутации в генах, кодирующих α-
(SCNN1A), β- (SCNN1B) и γ-субъединицы (SCNN1G) амилорид-
чувствительного натриевого канала, расположенные на хромосомах 12 (12p13) и 16 (16р13-р12).
При аутосомно-доминантных и спорадических формах заболевания причиной псевдогипоальдостеронизма является патология минералокортикоидного рецептора, ген которого расположен на коротком плече хромосомы 4 (4q31.1).
Отличительной особенностью клинической картины псевдогипоальдостеронизма, вызванного патологией натриевого канала, считают отсутствие поражения других минералокортикоидчувствительных тканей (потовые железы, кишечник). Лабораторно-диагностическими критериями данной патологии служит гиперкалиемия в сочетании с высоким содержанием альдостерона и ренина крови.
Минералокортикоиды в терапии псевдогипоальдостеронизма не эффективны, так как нарушен механизм самого действия альдостерона. Лечение таких пациентов сводится к возмещению потерь соли и воды.
Врождѐнная дисфункция коры надпочечников
Врождѐнная дисфункция коры надпочечников (адреногенитальный синдром, врождѐнная надпочечниковая гиперплазия) - группа заболеваний с аутосомнорецессивным типом наследования, в основе которых лежит дефект одного из энзимов или транспортных белков,
принимающих участие в биосинтезе кортизола в коре надпочечников. Снижение биосинтеза кортизола по принципу обратной связи приводит к повышению секреции АКТГ и, как следствие, к развитию гиперплазии коры надпочечников и накоплению метаболитов, предшествующих дефектному этапу стероидогенеза. В основе ферментативных нарушений лежат дефекты генов, кодирующих тот или иной фермент биосинтеза стероидов.
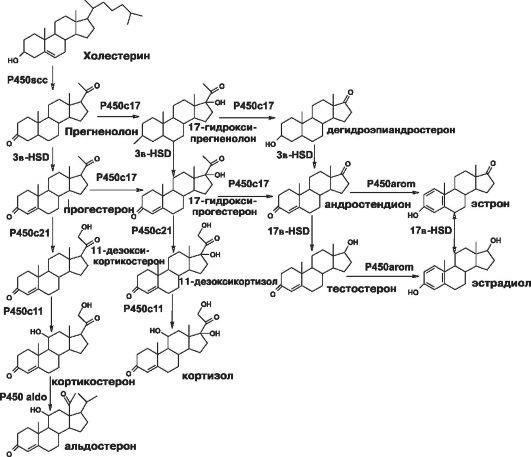
В зависимости от того, какой фермент стероидогенеза выпадает, различают 5 основных форм этого заболевания. Наиболее тяжѐлой формой считают липоидную гиперплазию надпочечников, обусловленную дефектом StARпротеина. При этой форме заболевания практически отсутствует синтез всех гормонов коры надпочечников, и ранее считали, что это несовместимо с жизнью. Наиболее часто диагностируют заболевание, обусловленное недостаточностью фермента 21-гидроксилазы. На долю этой формы приходится 75% всех случаев заболевания. Более редко наблюдают дефект 3-гидроксистероиддегид- рогеназы, недостаточность 17а-гидроксилазы или 11b-гидроксилазы. На рис. 18-5 представлена схема стероидогенеза. Клиническая картина заболевания зависит от места блока синтеза стероидов. Понятно, что будут наблюдать дефицит стероидов ниже блока и, наоборот, избыток стероидов, которые синтезируются до блока.
Недостаточность 21-гидроксилазы - одно из самых частых врождѐнных ферментативных нарушений стероидогенеза. Заболеваемость классическими вариантами болезни в различных популяциях колеблется от 1:10 000 до 1:18 000 новорождѐнных. Чрезвычайно высокая заболеваемость определена в двух изолированных популяциях: у эскимосов западной Аляски -1:280 и у
жителей острова Ла Руньон в Индийском океане - 1:2100. Заболевание наследуется по аутосомнорецессивному типу.
Этиология
Дефект 21-гидроксилазы обусловлен многочисленными мутациями гена, кодирующего этот фермент - CYP21. Ген расположен на коротком плече хромосомы 6.
Патогенез
21-гидроксилаза - микросомальный Р450-зависимый фермент, принимающий участие в биосинтезе кортизола и минералокортикоидов, трансформируя 17агидроксипрогестерон в 11-дезоксикортизол и прогестерон в дезоксикортикостерон. Недостаточность 21-гидрокси- лазы приводит к снижению продукции кортизола, что вызывает повышение секреции АКТГ и приводит к гиперплазии коры надпочечников. Надпочечники активно секретируют стероиды, предшествующие
Рис. 18-5. Схема стероидогенеза.
ферментативному блоку: 17а-гидроксипрогестерон и андрогены, биосинтез которых не зависит от 21-гидроксилазы.
Минералокортикоидную недостаточность различной степени выраженности диагностируют у 75% детей с 21-гидроксилазным дефицитом. Снижение концентраций дезоксикортикостерона и альдостерона приводит к
уменьшению реабсорбции натрия в почках. В связи с этим концентрация натрия в сыворотке крови падает и возрастает почечная реабсорбция калия. В результате этих нарушений развивается гипонатриемия, гиперкалиемия, ацидоз, обезвоживание. В ответ на снижение выработки минералокортикоидов возрастает рениновая активность плазмы.
Клиническая картина Пренатальная вирилизация
Классический вариант 21-гидроксилазной недостаточности приводит к выраженной гиперандрогении, формирующейся ещѐ внутриут-
робно. Предполагают, что активная вирилизация плода начинается с 20-25-й недели гестации, когда формируется влияние АКТГ на эмбриональный надпочечник и начинает синтезироваться кортизол. Внутриутробная гиперандрогения приводит к активной вирилизации наружных половых органов, что наиболее драматично выражено у девочек. К моменту рождения наружные половые органы девочки имеют бисексуальное строение: клитор гипертрофирован, отмечают сращение скротолабиального (мошоночного) шва различной степени выраженности. В некоторых случаях внутриутробная андрогенизация настолько выражена, что наружные половые органы практически соответствуют мужским, и девочку ошибочно регистрируют и воспитывают как мальчика (рис. 18-6 на вклейке).
Выделяют 5 степеней вирилизации наружных половых органов по Прадеру. Точкой отсчѐта считают нормальные наружные половые органы девочки (вирилизация отсутствует). Максимальная степень вирилизации соответствует нормальному строению наружных половых органов мальчика.
I степень вирилизации - гипертрофия клитора и нормальный вход
во влагалище.
II степень вирилизации - гипертрофия клитора и частичное сращение больших половых губ (высокая задняя спайка).
III степень вирилизации - клитор гипертрофирован и сформирована его головка, сращение половых губ формирует урогенитальный синус (единое мочеполовое отверстие у основания клитора).
IV-V степени вирилизации - гипертрофированный клитор напоминает нормальный половой член, однако наблюдают его искривление (фиксацию к промежности), урогенитальный синус открывается на стволе или головке полового члена (пинеальная уретра). Степень выраженности вирилизации у детей с недостаточностью 21-гидроксилазы значительно варьирует и обусловлена в первую очередь характером мутации гена CYP21. У мальчиков при рождении наружные половые органы соответствуют полу ребѐнка, может быть небольшое увеличение полового члена.
Постнатальная вирилизация
После рождения симптомы андрогенизации нарастают у детей обоего пола. У девочек увеличиваются размеры клитора, отмечают его напряжение. У мальчиков увеличиваются размеры полового члена, возникают эрекции. Следует отметить, что симптомы андрогенизации могут не проявляться в первые 1,5 года жизни ребѐнка. К 1,5-2 годам у детей обоего пола формируется половое оволосение, acne vulgaris, грубеет голос, гипертрофируется мускулатура. В первые годы жизни линейный рост детей ускорен, однако степень костной дифференцировки опережает рост. Зоны роста закрываются к 9-10 годам.
Степень пре- и постнатальной андрогенизации у пациентов с недостаточностью 21-гидроксилазы может иметь значительные индивидуальные колебания даже у больных сибсов в одной семье с одинаковым генетическим дефектом. Это может быть связано с индивидуальными особенностями метаболизма предшественников андрогенов и различием в активности рецепторов андрогенов у конкретного пациента.
Сольтеряющий синдром (синдром потери соли)
Полная потеря активности 21-гидроксилазы, наблюдаемая у 75% детей с дефицитом Р450с21, приводит к снижению биосинтеза альдостерона. Альдостерон необходим для поддержания нормального натриевого гомеостаза, а его дефицит приводит к потере натрия через почки, кишечник и потовые железы. Наличие выраженного сольтеряющего компонента, связанного с минералокортикоидной недостаточностью, представляет серьѐзную угрозу жизни ребѐнка с первых дней жизни. Через 3-4 дня после рождения нарастает гиперкалиемия, а спустя несколько дней развиваются гипонатриемия и гипернатриурия. Потеря соли приводит к выраженному обезвоживанию и снижению массы тела. Дегидратация усугубляется частой массивной рвотой, вызванной гиперкалиемией. При отсутствии терапии может наступить смерть ребѐнка в результате коллапса и кардиогенного шока.
Репродуктивная функция при классической форме недостаточности 21гидроксилазы
Пубертатный период у нелеченных детей обоего пола наступает поздно. У девочек даже при крайней степени вирилизации могут развиваться молочные железы (не более II степени по Таннеру) и появляться менструальные выделения. Регулярный менструальный цикл возможен только на фоне адекватной глюкокортикоидной терапии. Яичники уменьшены, с признаками поликистоза. Причины нарушения менструальной функции прежде всего обусловлены избыточной концентрацией надпочечниковых андрогенов, которые подавляют циклическую секрецию гонадотропинов и непосредственно угнетают развитие фолликула, вызывая его преждевременную атрезию.
У мальчиков функция гонад более сохранна, чем у девочек. У взрослых нелеченных пациентов возможна олигоспермия.
У детей обоего пола при поздно начатом лечении препаратами глюкортикоидов возможна преждевременная активация гипоталамо- гипофизарно-гонадной системы - истинное преждевременное половое развитие. Как правило, этот феномен наблюдают у детей, чей костный возраст к началу лечения достигает пубертатного: 11,5-12 лет у девочек и 13,5-14 лет у мальчиков (развитие сесамовидной кости). У девочек начинают увеличиваться молочные железы, у мальчиков увеличивается объѐм яичек. Причина ранней активации истинного пубертата у этих детей окончательно не ясна. Возможно, избыток по-
ловых стероидов изменяет чувствительность гипоталамических центров и способствует их «созреванию». Быстрое снижение избыточной секреции надпочечниковых андрогенов при назначении глюкортикоидной терапии способствует активации секреции гонадотропин-рилизинг гормона гипоталамусом, стимулирующего гонадотропную и гонадную функции. Ранний истинный пубертат у детей с недостаточностью 21-гидроксилазы ухудшает ростовой прогноз и требует присоединения антигонадотропной терапии.
Неклассическая форма недостаточности 21-гидроксилазы
Распространѐнность неклассических вариантов дефицита 21-гид- роксилазы в общей популяции очень высока и составляет до 0,3%. В некоторых этнических группах неклассическую форму заболевания наблюдают ещѐ чаще: 1,6% в Югославии, 1,9% в Испании, 3,7% у евреев Западной Европы (Ashkenazi). При неклассических формах заболевания снижение активности фермента 21гидроксилазы колеблется в достаточно широких пределах и может составлять 20-60% нормальных значений. В связи с этим клинические признаки гиперандрогении могут быть чрезвычайно вариабельны. Для детей с неклассической формой заболевания не характерны симптомы постнатальной вирилизации. При рождении наружные половые органы девочек сформированы по женскому типу. В редких случаях могут быть небольшое увеличение клитора и высокая задняя спайка на промежности, формирующая воронкообразный вход во влагалище. У детей обоего пола наиболее частым симптомом неклассической формы заболевания считают раннее появление лобкового и подмышечного оволосения (адренархе). Отмечают также небольшое увеличение скорости роста и костного созревания, однако конечный рост этих детей соответствует генетически ожидаемому.
У девочек пубертатного возраста и у взрослых женщин лѐгкая недостаточность 21-гидроксилазы проявляется в виде гирсутизма. Возможно нарушение менструальной функции и формирование поликистозных яичников, что приводит к бесплодию. Однако у 50% женщин с неклассической формой недостаточности 21-гидроксилазы репродуктивная функция не нарушена.
Определение уровня 17а-гидроксипрогестерона показано всем новорождѐнным, имеющим аномальное строение наружных половых органов при отсутствии пальпируемых яичек.
Параллельно проводят кариотипирование. Определение кариотипа 46ХХ у ребѐнка с бисексуальным строением наружных половых органов с 95%
вероятностью свидетельствует о наличии 21-гидроксилаз- ной недостаточности. Высокая концентрация 17а-гидроксипрогесте- рона окончательно подтверждает диагноз.
У недоношенных и детей, перенѐсших тяжѐлую родовую травму или рождѐнных с дефицитом массы тела при нормальных сроках гестации, содержание 17а-гидроксипрогестерона может быть повышено при отсутствии недостаточности 21-гидроксилазы. В этих случаях рекомендуют повторное проведение исследования (2-3-4 раза с интервалом 5-7 дней). Уменьшение содержания 17а-гидроксипрогестерона в динамике позволяет исключить 21-гидроксилазную недостаточность. Развитие сольтеряющего криза при недостаточности 21-гидроксилазы редко наблюдают у новорождѐнных и детей первых семи дней жизни. Однако до получения данных гормонального исследования, подтверждающих или исключающих 21-гидроксилазную недостаточность, всем детям необходимо проводить мониторинг уровня электролитов в крови.
Нарастание концентрации калия и снижение содержания натрия в сыворотке крови у ребѐнка с бисексуальным строением наружных половых органов, сопровождающиеся клиническими признаками сольтеряющего синдрома, следует рассматривать как проявление недостаточности 21-гидроксилазы и немедленно назначать терапию, не дожидаясь результатов гормонального анализа.
Наличие недостаточности 21-гидроксилазы у новорождѐнных мальчиков можно заподозрить лишь при наличии сольтеряющего синдрома. Всем новорождѐнным мальчикам с клиническими признаками гиперкалиемии, гипонатриемии и обезвоживания необходимо определять содержание 17агидроксипрогестерона.
Лабораторные и инструментальные исследования
Основной признак недостаточности 21-гидроксилазы - повышение уровня 17агидроксипрогестерона в сыворотке крови (в 10 раз и более) вследствие блока в синтезе кортизола. Необходимо исследование уровня калия и натрия в сыворотке крови для оценки степени минералокортикоидной недостаточности.
Содержание ренина крови значительно повышается при минералокортикоидной недостаточности.
У всех детей с бисексуальным строением наружных гениталий необходимо исследовать кариотип.
Молекулярная диагностика, основанная на |
определении |
мутаций в |
||
гене CYP21,позволяет точно подтвердить |
или исключить наличие |
|||
недостаточности |
21-гидроксилазы. |
Для |
пренатальной |
диагностики |
молекулярно-генетический метод считают единственно достоверным способом выявления заболевания и его формы у плода.
Диагностика и дифференциальная диагностика