

6 курс / Иммунология / Kazmirchuk_V_E_Kovalchuk_L_B_Maltsev_D_V_Klinicheskaya_immunologia
.pdf—ингибирование активности суперантигенов;
—изменение свойств Fc-рецепторов фагоцитов и вмешательство в Fc-onocpe- дованный фагоцитоз;
—ингибирование связывания комплемента и предотвращение образования МАК;
—конкуренция за распознавание антигена с растворимыми рецепторами лимфоцитов CD4 и CD8 и молекулами главного комплекса гистосовместимости II класса
—индукция транзиторной лимфопении, уменьшение уровня естественных киллеров и угнетение экспрессии LFA-1 на поверхности Т-лимфоцитов;
—угнетение функции CD8+ Т-лимфоцитов при помощи антител к свободному участку антигена главного комплекса гистосовместимости I класса.
Поскольку в основе заболевания лежит васкулит, оправдано использование сосудистых и антиагрегантных препаратов (трентал, курантил, ангинин, продектин, фенигидин и др.). Для улучшения качества жизни больных (купирования суставного болевого синдрома) применяют нестероидные противовос-
палительные средства.
§4. Иммунопатогенез, иммунодиагностика и иммунотерапия инсулинзависимого сахарного диабета
Инсулинзависимый сахарный диабет — это одно из наиболее распространённых органоспецифических аутоиммунных заболеваний. Различают три основные формы болезни: вирусиндуцированную, аутоиммунную и медленно прогрессирующую.
В первом случае развитие болезни связано с поражение островков Лангерганса различными вирусами, тропными к панкреатическим β-клеткам (острый вирусный панкреатит). Некоторые из них обладают прямым повреждающим действием (вирусы Коксакки, эпидемического паротита), другие же вызывают повреждение непрямым путём, способствуя срыву иммунологической толерантности к аутоантигенам β-клеток (вирусы герпес-группы). Известно, что сложные вирусы имеют мембраноподобную оболочку. Её образование происходит из модифицированных вирусом участков плазмолеммы клетки-хозяина. Поэтому считают, что срыв толерантности к β-Клеткам при вирусных инфекциях происходит по механизму молекулярной мимикрии — в условиях распознавания молекул вируса, комплексированных с клеточными поверхностными структурами, эффекторные иммунные реакции реализуются как против клеточных, так и против вирусных антигенов. Это означает, что поддержание аутоиммунной реакции возможно и после устранения вирусной инфекции.
Осуществление аутоиммунных реакций происходит за счёт накопления аутореактивных Т-хелперов 1 типа, т.е. иммунопатологический процесс при инсу-
линзависимом сахарном диабете разворачиваются преимущественно по клеточному типу. Собственно эффекторный механизм представлен специфическим и "неспецифическим"(по сути он не менее специфичен, чем первый) компонентами. Первый осуществляется за счёт деятельности аутореактивных цитотоксических Т-лимфоцитов, а второй — армированных макрофагов. Для обеспечения последних механизмом специфического распознавания панкреатических β-клеток (реакции антителозависимой клеточно-опосредованной цитотоксичности) необходимы аутоантитела, поэтому образуется определённое количество Т-хелперов 2 типа, способствующих пролиферации и созреванию аутореактивных В-лймфоцитов.
Вирусиндуцированной формой болезни чаще всего страдают лица мужского пола, дебют заболевания отмечается в молодом возрасте. Характерно наличие антигенов гистосовместимости В8, В12, DR3 и DR4.
Следует отметить, что инсулинзависимая форма сахарного диабета в связи с органоспецифичностью патологического процесса является самолимитирущимся аутоиммунным заболеванием — при полном уничтожении β-клеток аутоиммунные реакции прекращаются. В условиях вирусного поражения кинетика аутоиммунного процесса очень высока. Большинство клеток гибнут ещё в период инфицированное™, остальные разрушаются после устранения инфекционного начала по аутоиммунному механизму, разворачивающемся при посредничестве специфических аутоантител. На практике выявление таких антител затруднено, поскольку они существуют всего 3-4 месяца после устранения инфекционного начала — приблизительно такой срок необходим для полного завершения деструкции β-клеток панкреатических островков. В указанный период пациенты чувствуют себя удовлетворительно (так называемый медовый месяц — honey moon), поскольку эндокринная часть поджелудочной железы обладает огромным функциональным резервом и первые клинические проявления заболевания развиваются только после разрушения 80-90% β-клеток.
Аутоиммунная форма болезни протекает более медленно, аутоантитела к β-клеткам определяются на протяжении 1-2 лет. Причина срыва толерантао- с™ в этом случае недостаточно ясна, однако установлено, что она не связана с острыми вирусными панкреатитами. Нарушения толерантное™ при этой форме диабета более обширно, поэтому параллельно с аутоиммунным панкреатитом наблюдаются поражения других органов (тироидит Хашимото, аддисонова болезнь). Заболевание чаще встречается у женщин, дебют приходится на средний возраст. Наиболее характерны антигены гистосовместимоста HLA DR4, DR8, DRw3.
Медленно прогрессирующий инсулинзависимый сахарный диабета стартует поздно (в 40-50 лет), поэтому зачастую изначально таким пациентам ставится диагноз инсулиннезависимой формы болезни. Однако определение антител к β-клеткам панкреатических островков и выявление инсулинопении заставляют пересмотреть клинический диагноз. Указанная форма болезни встречается редко, причём наблюдается с одинаковой частотой среди мужчин и женщин. Характерного узкого спектра молекул гистосовместимости не установлено.
Клинические проявления. В клинике характерен синдром 5 "П", включающий полидипсию, полиурию, полифагию, поллакиурию и похудение. В тяжё-
лых случаях возможно развитие комы (кетоацидотического, гиперосмолярного или лактатацидемического генеза).
Лечение. Лечение состоит в пожизненном применении препаратов инсулина. Для лечения инсулинзависимого сахарного диабета были проведены апробации различных иммунотропных средств, однако убедительных положительных данных не было получено ни в одной из них.
§5. Иммунопатогенез, иммунодиагностика и иммунотерапия рассеянного склероза
Рассеянный склероз (PC) — аутоиммунное заболевание, в основе которого лежит демиелинизация волокон центральной нервной системы, проявляющаяся рассеянной в месте и времени органической симптоматикой. Этиология заболевания остаётся неизвестной. Предполагают, что срыв толерантности при PC происходит в результате вирусной нейроинфекции. При этом существуют две гипотезы, объясняющие связь между нейроинфекцией и развитием PC. Согласно первой, хроническая нейроинфекция (вирусы кори, простого герпеса, цитомегаловирус и др.), зачастую остающаяся субклинической, приводит к повышению проницаемости гематоэнцефалического барьера и изменению антигенного состава нервной ткани. Согласно другой, заболевание развивается в результате нарушения взаимоотношений в системе интегрированных вирусных генов и собственных генов нервных клеток. Известно, что около 5% генов клеток человеческого организма приобретены в результате интеграции нуклеиновых кислот различных вирусов в геном клеток-мишеней. Однако это не приводит к развитию аутоиммунных реакций, поскольку к вирусным интегрированным генам поддерживается иммунная толерантность. Нарушение такой толерантности может обусловить развитие аутоиммунной болезни. При этом заражение какой-либо инфекцией является необязательным, так как многие из интегрированных вирусных генов передаются из поколения в поколение.
Сегодня установлено, что в основе PC лежит синтез аутоантител к некоторым антигенам миелиновых оболочек нервной ткани. Наиболее изученными из них являются основной белок миелина, протеолипидный белок, миелинассоциированный гликопротеин, миелинолигодендроцитарный гликопротеин. На поздних стадиях заболевания синтезируются аутоантитела к антигенам .-нейронов и клеток нейроглии. Причиной продукции таких аутоантител является активация специфических аутореактивных Т-хелперов 1 типа. Доступ антител к антигенам миелина обеспечивается за счёт повышения проницаемости гематоэнцефалического барьера под влиянием провоспалительных медиаторов — ИФН-γ, ΦΗΟ-α, ΦΗΟ-β, ИЛ-2, которые, как известно, относятся к комплекту цитокинов Th 1. В многочисленных исследованиях показано, что повышение уровня этих цитокинов коррелирует с нарастанием клинической симптоматики и ухудшением состояния пациента. Наоборот, повышение уровня цитокинов Th

2 (ИЛ-4, ИЛ-5, ИЛ-6 и ИЛ-10) сопровождается стиханием патологического процесса и возникновением клинической ремиссии заболевания. Однако следует отдавать себе отчёт, что причиной указанных изменений является не собственно цитокины, а те иммунные процессы, отражением которых являются изменения цитокинового баланса. Так как в нервной системе нет лимфоидной ткани, поначалу для развития воспалительного процесса необходима миграция аутореактивных лимфоцитов извне, однако в последующем синтез провоспалительных цитокинов начинают осуществлять клетки нейроглии, относящиеся к компонентам врождённой резистентности организма. Таким образом, на поздних стадиях патологический процесс во многом обеспечивается за счёт ресурсов самой нервной системы. Эффекторным звеном при PC являются армированные макрофаги, т.е. осуществляются реакции антителозависимой клеточно-опосредованной цитотоксичности.
Морфологическим субстратом болезни являются очаги демиелинизии нервных волокон (белого вещества) центральной нервной системы (так называемые бляшки). В условиях ремиссии отмечается ремиелинизация за счёт деятельности олигодендроцитов (шванновских клеток). По мере прогрессирования болезни происходит не только разрушение миелина, но и повреждение шванновских клеток (гипоксия, ацидоз, действие свободных радикалов), что замедляет процесс ремиелинизации и способствует нарастанию резидуального дефекта, который не устраняется даже во время ремиссии. Замедление ремиелинизации в результате гибели олигодендроцитов в очагах воспаления приводит к ещё одному трагическому последствию. Дело в том, что миелиновая оболочка защищает аксональные цилиндры, приимая на себя "удар" агрессивных агентов (свободных радикалов, токсических перекисей, лизосомальных ферментов). При нарушении ремиелинизации происходит повреждение собственно нервных структур, что приводит к необратимым изменениям и формированию астроглиальных рубцов в месте повреждений за счёт компенсаторной пролиферации астро— и глиоцитов. Указанные механизмы лежат в основе трансформации ремиттирующего течения болезни во вторично прогрессирующее.
Клиника. Основные клинические синдромы PC представлены в табл. 47.
Таблица 47. Основные клинические синдромы PC
1.Синдром поражения пирамидного тракта
2.Синдром поражения путей мозжечка
3.Синдром поражения черепно-мозговых нервов
4.Синдром чувствительных расстройств
5.Нарушение функции тазовых органов
6.Зрительные нарушения
7.Нейропсихологические нарушения
Лабораторная диагностика. Для подтверждения диагноза рассеянного склероза помимо характерных результатов нейровизуализационных методов (например, магнитно-резонансной томографии мозга) необходимо выявление высоких

|
титров аутоантител к основному бел- |
|||||
|
ку миелина в сыворотке крови и лик- |
|||||
|
воре. |
При |
анализе |
иммунограммы |
||
|
можно выявить признаки активации |
|||||
|
иммунной системы преимуществен- |
|||||
|
но за счёт клеточного звена — повы- |
|||||
|
шение количества Т-хелперов, воз- |
|||||
|
растание |
иммунорегуляторного ин- |
||||
|
декса, высокие концентрации цито- |
|||||
|
кинов ΦΗΟ-α, ИЛ-2, ИФН-γ. Часто |
|||||
|
имеет место низкое количество регу- |
|||||
|
ляторных Т-клеток и малая концен- |
|||||
|
трация интерлейкина 10 в сыворотке |
|||||
|
крови. |
|
|
|
|
|
|
При |
анализе спинномозговой |
||||
|
жидкости методом |
изоэлектричес- |
||||
|
кого |
фокусирования |
можно выявить |
|||
|
характерные |
олигоклональные |
поло- |
|||
|
сы иммуноглобулинов (рис. 54). Сы- |
|||||
|
вороточные антитела к аквапорину 4 |
|||||
|
(NMO-IgG), водному каналу астроци- |
|||||
|
тов, помогают отличить нейромиелит |
|||||
|
зрительного нерва от рецидивирующе- |
|||||
|
ремитгирующего PC с чувствительно- |
|||||
|
стью 73% и специфичностью 91%. |
|||||
Рис. 54. Олигоклональные полосы имму- |
Целесообразна также диагности- |
|||||
ноглобулинов цереброспинальной жидкости у |
ка персистирующих |
нейротропных |
||||
больного с рассеяным склерозом |
вирусов |
(ДНК-гибридизация, |
ПЦР |
|||
|
и др.), учитывая их возможную роль |
|||||
в инициации и поддержании аутоиммунного процесса при PC.
Лечение. При обострении заболевания используют препараты глюкокортикоидов и АКТГ, а также плазмаферез. Иногда может быть полезной внутривенная иммуноглобулинотерапия в дозе 2 г/кг в течение 2-5 дней. Предупреждение последующих обострений достигается за счёт интерферонотерапии и применения глатирамер ацетата (копаксона).
Наиболее эффективными для лечения ремиттирующей и вторично прогрессирующей формы болезни оказались препараты интерферона-β. Иммунологические аспекты их терапевтического воздействия связаны со снижением продукции ИФН-γ и ΦΗΟ-α, а также с изменением экспрессии антигенпредставляющих и адгезионных молекул. Бетаферон (интерферон β-lb) вводят п/ кж в дозе 8 млн ЕД через день. Авонекс (интерферон β-1а) используют в дозе 6 млн ЕД в/м 1 раз в неделю. Ребиф (интерферон β-1а) вводят 3 раза в неделю по 12 млн ЕД п/кж. Длительность лечения составляет месяцы и годы, а его эффективность достигает 75%. Основной проблемой интерферонотерапии PC является плохая переносимость препаратов, которые вызывают гриппоподоб-
ный синдром, повышение артериального и внутричерепного давления, усиление судорожной готовности.
Копаксон (глатирамер ацетат) является синтетическим полимером, состоящим из четырёх аминокислот: L-глутамина, L-лизина, L-аланина, L-тирозина. По своему составу копаксон подобен иммуногенным пептидам основного белка миелина — одного из главных аутоантигенов при PC. Механизм действия препарата предположительно связан с конкуренцией с причинными иммуногенным и пептидами за антигенпредставляющие рецепторы (молекулы гистосовместимости II класса). Копаксон используют в дозе 20 мг ежедневно п/кж. В целом эффективность лечения копаксоном ниже, чем интерферонотерапии (около 30%), однако отмечается лучшая переносимость лечения.
Имеются сообщения об эффективности при PC натализумаба, представляющего собой гуманизированные моноклональные антитела к а4-интегрину, которые блокируют адгезию мононуклеарных клеток периферической крови к эндотелию, снижая миграцию клеток через ге матоэ н цефал и чес к и й барьер. Проходит клинические испытания другой иммунотропный препарат для профилактического лечения PC — финголимод, являющийся модулятором сфингозин-1-фосфата, что приводит к изоляции лимфоцитов в периферических иммунных органах, снижению их циркуляции и миграции.
В последние годы появились серьёзные научные работы российских учёных, которые демонстрируют эффективность профилактического лечения PC амиксином — индуктором синтеза эндогенного интерферона. Препарат применяется во вторую половину дня 3 раза в неделю по одной таблетке на протяжении нескольких месяцев или лет. Также имеются доказательства эффективности препарата галавит, оказывающего модулирующее действие на клеточное звено врождённого иммунитета.
У больных PC необходимо активно выявлять и лечить хроническую герпетическую инфекцию, что может существенно снизить частоту и тяжесть рецидивов болезни.
§6. Иммунопатогенез, иммунодиагностика и иммунотерапия миастении гравис
Миастения гравис — классическое заболевание аутоиммунной природы, развитие которого обусловлено повреждением двигательных конечных пластинок поперечнополосатой мускулатуры вследствие взаимодействия аутоантител с Н-холинорецепторами, клинически проявляющееся в виде прогрессирующей мышечной слабости.
Исторические аспекты. Заболевание известно научному миру со середины 17 века благодаря описанию Т. Willis. Такое раннее вимание к болезни обусловлено тем, что миастения являлась непосредственной причиной смерти в связи с развитием паралича дыхательных мышц. На протяжении продолжи-
тельного времени механизм болезни оставался неизвестным. Лишь в 1960 году D. Simpson высказал предположение об аутоиммунной природе заболевания и рассматривал его как полимиозит, ограниченный конечными пластинками.
Существенную роль в поимании патогенетических механизмов миастении сыграло исследование яда змей и выявление α-bungarotoxin, способного Избирательно взаимодействовать со структурными компонентами Н-холинорецепторов постсинаптических мембран. При этом активные центры рецепторов остаются интактными. Тем не менее, подобное взаимодействие приводит к конформационной перестройке молекулы, сопровождающейся потерей чувствительности активных центров к медиатору. В соответствии с современными взглядами, на структурных компонентах Н-холинорецепторов (Н-Хр) конечных пластинок содержатся антигенные детерминанты, взаимодействие которых с Fab-фрагментами аутоантител вызывает пространственную перестройку макромолекулы рецептора, затрагивающую и центры связывания ацетилхолина (АЦХ). Это обуславливает снижение чувствительности постсинаптической мембраны (ПостСМ) к медиатору и нарушение синаптической передачи.
Физиология синаптической передачи. Мионевральный синапс является высокоспециализированной системой, состоящей из пресинаптической мембраны (ПреСМ), синаптической щели (СЩ) и постсинаптической мембраны (ПостСМ). Последняя имеет складчатое строение и содержит Н-Хр. Н-Хр являются гликопротеинами с молекулярной массой 258000 Д и состоят из 5 субъединиц (2-α, β, γ и δ), формирующих центральный канал, закрытый в спокойном состоянии. Работа синапса объясняется квантово-везикулярной теорией. Медиатор АЦХ накапливается в терминали аксона в специальных везикулах, которые в глубине аксона располагаются хаотично, а ближе к ПреСМ образуют группировки, именуемые кластерами. Каждый кластер расположен напротив активных мембранных зон, из которых возможен экзоцитоз ацетилхолина. Во время поступления нервного импульса осуществляется слияние мембран везикул кластеров с ПреСМ, вследствие чего происходит высвобождение ацетилхолина в синаптическую щель. Дальнейшая судьба медиатора связана с диффузией его по направлению к ПостСМ, где концентрация АЦХ практически равняется нулю за счет деятельности фермента ацетилхолинэстеразы (АЦХЭ), способного быстро расщеплять медиатор до холина и уксусной кислоты. Каждый из образованных компонентов не обладает самостоятельной медиаторной активностью. Закономерность направленного перемещения АЦХ справедлива лишь для всего пула высвобожденного медиатора, предусмотреть же поведение отдельной молекулы можно лишь с определенной степеньюгвероятности. Диффузионная траектория движения АЦХ существенно отличается от прямой и является особенной у каждой молекулы, однако за счет небольшой дистанции пробега (глубина СЩ) все молекулы АЦХ прибывают на ПостСМ почти одновременно. Это имеет большое значение для синаптической передачи, поскольку одиночные, разрозненные молекулы имеют повышенный риск быть расщепленными АЦХЭ. При взаимодейсвтии ацетилхолина с Н-Хр ионные каналы внутри молекул рецепторов открываются, и благодаря поступлению ионов Na+ в саркоплазму, возникает местная деполяризация ПостСМ — пост-
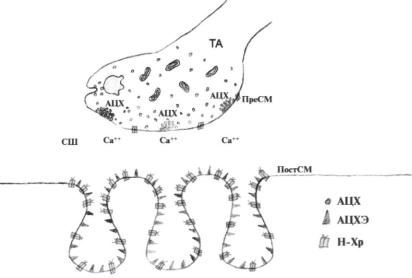
синаптический потенциал. Если величина вызванного постсинаптического потенциала превышает пороговый уровень, то возбуждение генерализуется — формируется потенциал действия, который в виде волны деполяризации распространяется вдоль сарколеммы и в дальнейшем углубляется в саркоплазму по трубочкам, обеспечивая сокращение мышцы.
Наличие субнейральных складок и углублений существенно увеличивает площадь рецепторного поля, тем не менее это явление нельзя трактовать однозначно. Дело в том, что Н-Хр распределены вдоль ПостСМ крайне неравномерно. Подавляющее большинство рецепторов содержится на гребнях субнейральных складок (точнее, в участках их склонов, обращенных к противоположной складке) в то время, как вдоль углублений расположены одиночные Н-Хр. Кроме того, для сокращения мышцы достаточным является возбуждение лишь 30-40% имеющихся рецепторов.
В связи с этим считают, что значимо функционально активной является не вся ПостСМ, а лишь своеобразные координационные центры, морфологическим субстратом которых являются склоны гребней смежных складок, обращенные друг к другу. Они имеют 3 важных с точки зрения синаптической передачи свойства:
1)сюда поступает максимальное количество АЦХ, поскольку данные участки расположены точно напротив активных центров ПреСМ;
2)именно здесь самая большая плотность расположения рецепторов;
3)здесь достигается синхронизация поступления медиатора, благодаря малой глубине СЩ (рис. 55).
Рис. 55. Мионевральный синапс в покое
Все это обуславливает ослабление АЦХЭ барьера, а, значит, наиболее точную координацию между импульсацией, поступающей на терминаль аксона,
и эффектом — сокращением мышцы. Всегда есть возможность расширения площади таких центров в глубину складок, то есть, они являются динамическими. Все эти данные позволяют сделать важный вывод, что архитектоника ПостСМ не менее значима, чем количество АЦХ или функционирующих Н-Хр (рис. 56).
Патоморфология миастенического синапса. Миастения относится к разряду
органоспецифических аутоиммунных болезней. При этом иммунопатологические процессы, имеющие непосредственное клиническое проявление, разворачиваются в мионевральном синапсе, вследствие чего последний приобретает специфические черты, являющиеся сочетанием собственно патологических и компенсаторных изменений. Классический миастенический синапс характеризуется следующими признаками:
1)уменьшением высоты субнейральных складок, их нерегулярностью, сглаженностью рельефа ПостСМ;
2)увеличением глубины СЩ;
3)компенсаторным повышением запасов АЦХ и активности холинацетилтрансферазы в терминале аксона;
4)увеличением площади ПостСМ, вызванным компенсаторным новообразованием терминалей аксонов, однако новые синаптические зоны формируются с первично нарушенной складчатостью (рис. 57).
Дело в том, что при формировании новых синаптических зон в постнатальном периоде онтогенеза не происходит надлежащего погружения терминали аксона в мышечное волокно. Следует учитывать и тот факт, что сложный процесс формирования новых синаптических зон происходит в условиях их постоянного аутоиммунного повреждения.
Иммунопатогенез. Для объяснения данных утверждений рассмотрим иммунопатологические процессы, которые происходят в синапсе при миастении. Считают, что антигенные детерминанты содержатся в α-субъединицах Н-Хр. При этом аутоантитела (в основном IgG3) оказывают разный эффект на Н-Хр. Некоторые из них являются блокирующими и при взаимодействии с рецептором делают невозможным связывание последнего с АЦХ, хотя рецептор остается на поверхности ПостСМ. Предполагают, что для полной блокады необходимо взаимодействие, по крайней мере, с несколькими аутоАТ. Возможно, это связано с наличием нескольких центров связывания АЦХ на одной молекуле рецептора. Кроме того, была отмечена усиленная деградация Н-Хр, связанных с аутоАТ.
Известно, что для ускоренного разрушения рецепторов необходимо перекрестное связывание их бивалентными IgG. Несмотря на усиленную деградацию, скорость ресинтеза Н-Хр остается неизменной, что приводит к прогрессирующему уменьшению их количества на ПостСМ. И, наконец, третьим результатом взаимодействия Н-Хр с аутоАТ является присоединение к Fcфрагментам последних С1 компонента комплемента с последующим вовлечением всего каскада, что сопровождается повреждением как самого рецептора (фракции комплемента обладают ферментативной активностью), так и ПостСМ за счет формирования мембранатакующих комплексов и цитотоксического действия макрофагов.

Рис. 56. Мионевральный синапс в процессе возбуждения
Рис. 57. Мионевральный синапс при миастении
Генерализованного повреждения собствено мышечных волокон не происходит, поскольку площадь ПостСМ несоразмеримо мала по сравнению с размером волокна. Доказательством высвобождения из конечной пластинки фрагментов мембраны, содержащих Н-Хр, является выявление комплексов