

Синдром бабочки: буллёзный эпидермолиз
Введение:
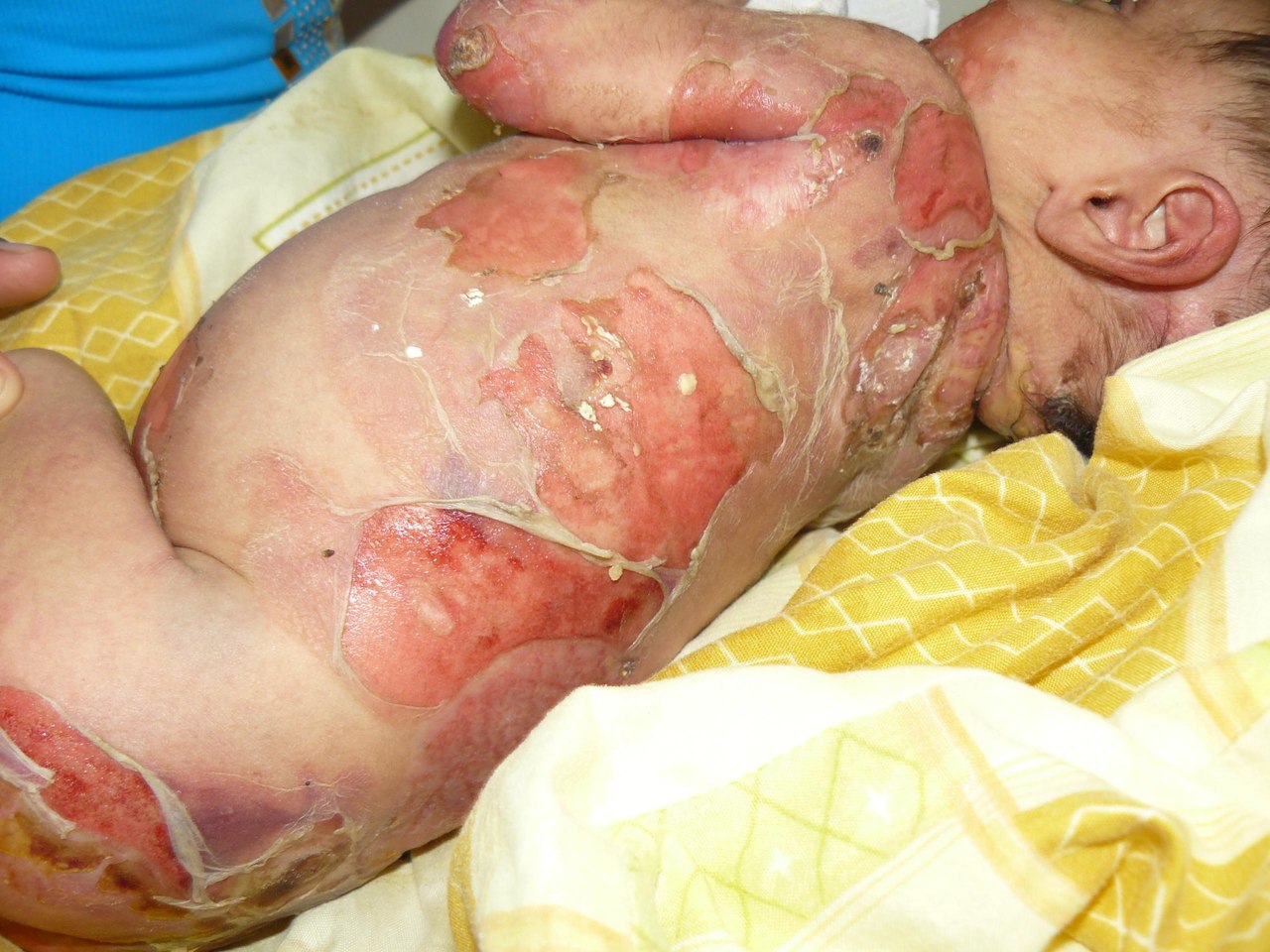
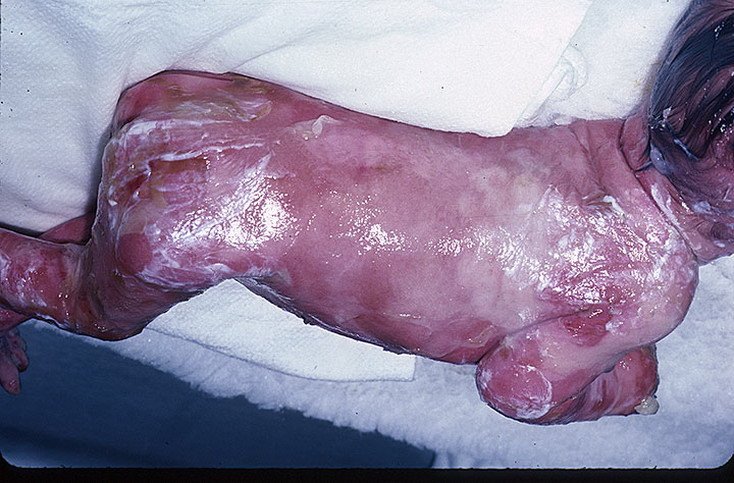
Буллезный эпидермолиз (БЭ), или, как его еще называют, синдром бабочки – это очень редкое наследственное заболевание. Таких детей прозвали детьми-бабочками. Если с этим названием у вас возникают какие-то красивые ассоциации, то сама болезнь далеко не эстетична. На самом деле, больные буллезным эпидермолизом всю свою жизнь проводят в мучительных страданиях. Причина - их кожа, хрупкая, как крылья бабочек.

К сожалению, болезнь «синдром бабочки» вылечить невозможно. Потому что она глубоко заложена на генетическом уровне. Таким детям необходим специальный уход: полностью мягкое окружение, одежда и обувь. В России эта неизлечимая болезнь даже не внесена в список самых редких заболеваний. Однако существует специализированный фонд помощи детям-бабочкам «БЭЛА» Заболевание «синдром бабочки» не известно широкой публике. Мало кто даже может выговорить ее сложное название – буллезный эпидермолиз. Однако такие дети есть по всей России и в странах СНГ. У каждого из больных одинаковая проблема: местные врачи не знают, как с ними поступить, диагноз ставится "на глаз", и не всегда верный относительно видов этого заболевания. А порой матери вообще прячут таких детей, что приводит к их гибели из-за неправильного ухода.
Синдром бабочки – это результат неправильного формирования белка в коже или полное его отсутствие. Также причиной развития болезни могут стать спонтанные генные мутации.
Синдром бабочки характеризуется появлением на коже человека пузырьковой сыпи. При рождении кожа у больного ребенка сползает как шкурка у змеи. Конечно же, это вызывает шоковую реакцию у матери. Многие просто отказываются от больных детей еще в родильном доме. Но шанс выжить у ребенка с болезнью буллезный эпидермолиз в детском доме практически равен нулю.
История:
Термин «буллёзный эпидермолиз» впервые был использован в 1886 г. немецким дерматологом Генрихом Кёбнером, хотя случаи, схожие с данным диагнозом, описывались и до него. В 1962 г. Пирсоном была разработана первая схема классификации БЭ, основанная на применении трансмиссионной электронной микроскопии. Были выделены три основных типа: эпидермолитический, люцидолический и дермолитический, исходя из уровня образования пузырей на ультраструктурном уровне. В 1980-х годах была разработана техника иммунофлюоресцентного исследования образцов кожи, расширенная впоследствии иммуногистологическими методами (иммуногистохимией и иммунофлюоресценцией). С 1990-х годов почти для каждого подтипа БЭ были определены мутации в конкретных генах структурных белков кожи, которые изменяются при БЭ.
Классификация:
Исторически сложилось так, что буллёзный эпидермолиз был классифицирован в соответствии с морфологией кожи. Наследственные варианты БЭ в настоящее время разделяются на три большие группы в соответствии с уровнем образования пузырей в тканях, отдельно выделен синдром Киндлера: простой буллезный эпидермолиз (ПБЭ), пограничный буллезный эпидермолиз (ПоБЭ), дистрофический буллезный эпидермолиз (ДБЭ), синдром Киндлера (разный уровень образования пузырей).
Основной тип БЭ |
Основные подтипы БЭ |
Белки-мишени |
Простой БЭ (ПБЭ) |
Супрабазальный ПБЭ |
плакофилин-1; десмоплактин; возможно другие |
|
Базальный ПБЭ |
α6β4-интегрин |
Пограничный БЭ (ПоБЭ) |
ПоБЭ, подтип Херлитца |
ламинин-332 (ламинин-5) |
|
ПоБЭ, другие |
ламинин-332; коллаген XVII типа; α6β4-интегрин |
Дистрофический БЭ (ДБЭ) |
доминантный ДБЭ |
коллаген VII типа |
|
рецессивный ДБЭ |
коллаген VII типа |
Синдром Киндлера |
- |
киндлин-1 |
Эпидемиология:
Частота встречаемости БЭ варьирует от 1:30000 до 1:100000 человек. По данным Национального регистра БЭ (США), распространенность всех типов БЭ составляет 8,22:1 млн. человек. Распространенность ПБЭ – 10,75 на 1 000 000, ПоБЭ – 2,04 на 1 000 000, ДДБЭ – 2,86 и на 1 000 000, РДБЭ — 2,04 на 1 000 000 человек.
Генетика:
БЭ наследуется по аутосомно-доминантному и аутосомно-рецессивному типу. При БЭ мутации обнаруживаются в более чем 10 генах. Описаны различные виды мутаций – мисенс-мутации, нонсенс-мутации, делеции, мутации рамки считывания, инсерции, мутации сайта сплайсинга. При наиболее распространенных подтипах ПБЭ обнаруживаются мутации в генах KRT5 и KRT14, примерно в 75% случаев, при этом вероятно, что мутации в других пока неидентифицированных генах могут также вызывать развитие ПБЭ. При ПоБЭ чаще всего мутации обнаруживаются в генах LAMB3 (70% случаев), LAMA3, LAMC2, COL17A1. В большинстве случаев заболевание наследуется по аутосомно-рецессивному типу, однако, описаны случаи соматического мозаицизма и однородительской дисомии. При ДБЭ описаны мутации в гене COL7A1, в 95% случаев доминантного и рецессивного типов БЭ.