

В почках: из аргинина и глицина образуется гуанидинацетат.
Гуанидинацетат транспортируется в печень.
В печени: происходит метилирование гуанидинацетата при участии SAM и образуется креатин.
Креатин с током крови транспортируется в мышцы и клетки мозга, где из него образуется соединение с макроэргической связью – креатинфосфат.
Реакция синтеза креатинфосфата легко обратима.
Креатинфосфат накапливается в мышечной и нервной тканях и служит резервной формой АТФ.
! В работающей мышце первые секунды работы происходит реакция расщепления креатинфосфата с образованием АТФ при участии креатинкиназы (субстратное фосфорилирование).
Чуть позже включается анаэробный гликолиз.
И только при дальнейшей работе включаются β-
окисление жирных кислот и аэробный распад глюкозы.
В мышцах: часть креатинфосфата неферментативно превращается в креатинин.
Креатинин не используется клетками и выводится с мочой.
В норме: с мочой выделяется ~ 1-2 г креатинина в сутки.
Определение содержания креатинина в моче используется для характеристики клубочковой фильтрации и диагностики заболеваний почек.
Регенерация метионина
В клетках организма происходит очень большой расход метионина, т.к. реакции трансметилирования происходят очень интенсивно.
Т.к. метионин – незаменимая аминок-та, то большое значение имеет регенерация метионина при участии заменимых аминокислот серина и глицина:
1) В ходе реакций трансметилирования от SAM отщепляется CH3-группа и он превращается в
SAГ:
R + SAM–S+–CH3 → R–CH3 + SAГ

2) SAГ под действием гидролазы распадается на аденозин и гомоцистеин:
SH
CH2
SAГ + H2O  Аденозин + CH2
Аденозин + CH2
HC NH2
COOH
Гомоцистеин
3) Гомоцистеин может превращаться в метионин при участии фермента гомоцистеинметилтрансферазы.
|
|
|
|
|
|
|
Метилен-H4- |
|
|
CH3 |
|||
|
|
|
|
|
|
|
-фолат |
|
|
||||
|
|
|
|
|
|
|
|
||||||
SH |
|
|
|
Гли |
|
|
|
||||||
|
|
|
S |
||||||||||
|
|
CH2 |
Метил-H4- |
|
Сер |
|
|
CH2 |
|||||
|
|
|
|||||||||||
|
|
|
|
|
|||||||||
|
|
|
|
||||||||||
|
|
CH2 |
-фолат |
H4-фолат |
|
|
CH2 |
||||||
|
|||||||||||||
|
|
|
|||||||||||
HC |
|
NH2 |
|
|
|
|
HC |
|
NH2 |
||||
|
|
B12 |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
COOH |
|
|
|
COOH |
|||||||||
|
|
|
|
||||||||||
Гомоцистеин |
|
|
Метионин |
||||||||||
Донор метильной группы в этой реакции – метил- H4-фолат.
Промежуточный переносчик метильной группы – метилкобаламин (производное витамина B12).
4) Образующийся метионин может снова активироваться и повторно использоваться в реакциях трансметилирования:
Метионин + АТФ → SAM + PPi + Pi

Обмен Фенилаланина и Тирозина
Фенилаланин – незаменимая аминокислота.
Используется в организме только в 2-х процессах: Субстрат для синтеза белков и превращается в тирозин.
! Превращение Фен в Тир необходимо в 1 очередь для удаления избытка Фен, т.к. его высокие концентрации токсичны для клеток.
В Тир превращается ~ 90% Фен:
Превращение Фен в Тир – это 1-ая реакция основного пути метаболизма Фен.
Все дальнейшие превращения в организме происходят уже с Тир.
При нарушении протекания реакции превращения Фен в Тир возникает заболевание – фенилкетонурия (фенилпировиноградная олигофрения).
Из-за невозможности превращения Фен в Тир, катаболизм фенилаланина протекает по
альтернативному пути:
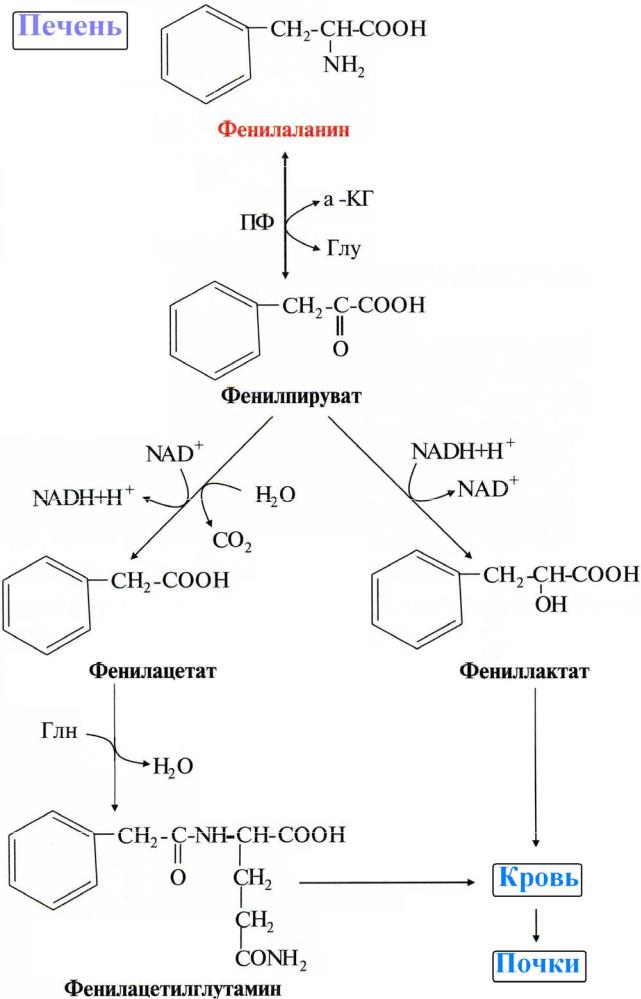
При этом в крови и моче повышается содержание фенилаланина и метаболитов альтернативного пути: фенилпирувата, фениллактата, фенилацетата и др.
Симптомы: Резкое нарушение умственного и физического развития, судорожный синдром, «мышиный» запах.
Также встречается: нарушение пигментации кожи.
Фен и его производные, при их избытке, оказывают токсическое действие на клетки мозга, поскольку:
ограничивают транспорт Тир и Три через гематоэнцефалический барьер и тормозят синтез нейромедиаторов (дофамина, норадреналина, серотонина).
Без лечения больные фенилкетонурией не доживают до 30 лет.
Заболевание наследуется по аутосомнорецессивному типу.
Выделяют 2 формы фенилкетонурии:
1) Классическая фенилкетонурия:
Причина: наследственный дефект фермента фенилаланингидроксилазы.
Частота заболевания: 1 случай на ~ 10000 новорожденных.
2) Вариантная фенилкетонурия: (коферментзависимая гиперфенилаланинемия)
Причина: мутации в генах, контролирующих метаболизм H4-биоптерина.
Встречается: 1-2 случая на ~ 1 млн. новорожденных.
H4-биоптерин необходим для гидроксилирования не только Фен, но и Тир и Три, поэтому, при этой форме заболевания нарушен метаболизм всех 3 аминокислот, а также синтез многих нейромедиаторов.
При этой форме заболевания возникают тяжелые неврологические нарушения и ранняя смерть.