

Постишемическое повреждение головного мозга!
.pdfMINIREVIEW
Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy
Giuseppe Pignataro, Antonella Scorziello, Gianfranco Di Renzo and Lucio Annunziato
Division of Pharmacology, Department of Neuroscience, School of Medicine, ‘‘Federico II’’ University of Naples, Italy
Keywords
cerebral ischemia; ionic homeostasis; kinase; MnSOD; neuroprotection; nitric oxide; postconditioning; preconditioning; stroke; tolerance
Correspondence
L. Annunziato, Division of Pharmacology, Department of Neuroscience, School of Medicine, ‘‘Federico II’’ University of Naples, Naples, Italy
Fax: +39 081 7463323
Tel: +39 081 7463318 E-mail: lannunzi@unina.it
(Received 30 July 2008, revised 4 September 2008, accepted 13 October 2008)
doi:10.1111/j.1742-4658.2008.06769.x
Because clinical trials of pharmacological neuroprotective strategies in stroke have been disappointing, attention has turned to the brain’s own endogenous strategies for neuroprotection. Two endogenous mechanisms have been characterized so far, namely ischemic preconditioning and ischemic postconditioning. The neuroprotective concept of preconditioning is based on the observation that a brief, noninjurious episode of ischemia is able to protect the brain from a subsequent longer ischemic insult. Recently, a hypothesis has been offered that modified reperfusion subsequent to a prolonged ischemic episode may also confer ischemic neuroprotection, a phenomenon termed postconditioning. Many pathways have been proposed as plausible mechanisms to explain the neuroprotection offered by preconditioning and postconditioning. Unfortunately, so far, none of them has clearly identified the mechanism involved in preconditioning and postconditioning. The present article will review the main mechanisms reported to date to explain the neuroprotective effect of both ischemic preconditioning and postconditioning.
Stroke is the third most common cause of death after heart attack and cancer and has profound negative social and economic effects. The failure, over the past three decades, of multiple clinical trials of exogenously administered drugs as potential stroke neuroprotectants has enhanced the ongoing search to identify endogenously modulated mechanisms activated after cerebral ischemia that might be harnessed as neuroprotectants in stroke [1–3]. Such molecular mechanisms might be those evolutionarily conserved to counteract the damage induced by a disruption to the cerebral blood supply [4].
In this attempt, ischemic preconditioning and ischemic postconditioning represent two promising strategies in modulating ischemic damage. The neuroprotective concept of preconditioning is based on the observation that a brief noninjurious episode of ischemia is able to protect the brain from a subsequent longer ischemic insult [1]. A major goal of this research is to understand the mechanisms involved in the preconditioning-induced neuroprotection in an attempt to identify genes and proteins increased in abundance during preconditioning as potential drug targets for stroke therapy. In fact, such preconditioning as a strategy to attenuate the patho-
Abbreviations
APC, anoxic preconditioning; ASIC1a, acid-sensing ionic channel 1a; CCA, common carotid artery; eNOS, endothelial nitric oxide synthase; ERK, extracellular regulated kinase; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; JNK, c-jun N-terminal kinase; MAPK, mitogenactivated protein kinase; Mn-SOD, manganese–superoxide dismutase; MPTP, mitochondrial permeability transitional pore; NCX, sodium calcium exchanger; NHE, Na+ ⁄ H+ exchanger isoform 1; NKCC1, Na+ ⁄ K+2Cl) cotransporter isoform 1; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PMCA, plasma membrane calcium ATPase; TNF-a, tumor necrosis factor-alfa.
46 |
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
G. Pignataro et al. |
Focus on preconditioning and postconditioning |
physiological consequences of ischemia–reperfusion injury would be focused on pretreatment situations, such as protection before cardiac bypass surgery. A nonpharmacological neuroprotective strategy for administration after ischemia onset, however, remains elusive.
Based on recent studies on the heart [5–7] and proof- of-principle experiments in the brain [8–10], a hypothesis has been offered that a modified reperfusion subsequent to a prolonged ischemic episode may also confer ischemic neuroprotection, a phenomenon termed postconditioning [10,11]. In published studies of the myocardium describing postconditioning, a number of issues are presented. Yellon has suggested that preconditioning and postconditioning are similar phenomena, with the effectors being a similar group of downstream signalling cascades [7]. By contrast, the mechanisms regulating postconditioning might be entirely different from those regulating preconditioning, as the rapidity of onset of postconditioning-induced neuroprotection contrasts with a significant temporal delay for (protein synthesis-dependent) preconditioning-induced neuroprotection. Furthermore, postconditioning may not involve the activation of endogenous neuroprotection, but rather merely attenuate the burst of free radicals occurring with reperfusion. Indeed, it has been suggested that the protection of postconditioning could be accomplished by a gradual increase in the reperfusion rate [12,13]. Other hypotheses have also been offered, such as postconditioning resulting from intravascular adenosine washout [14] or effected by intravascular pressures associated with reperfusion [15]. However, the major focus of postconditioning mechanisms is the role of the effector protein kinases and the question of the similarity or difference of these effectors versus those thought to be involved in preconditioning [5,6]. Although the time-window of effectiveness of postcon- ditioning-induced neuroprotection is narrow, postconditioning may have translational relevance to reperfusion and thrombolitic treatments in acute brain ischemia.
Over the last 5 years, some exhaustive and basic reviews addressing the mechanisms involved in ischemic preconditioning have been published [1–3], whereas reviews covering the main aspects involved in the neuroprotection mediated by postconditioning are still lacking. Therefore, the present article will review the main mechanisms reported to date to explain the neuroprotective effect of both ischemic preconditioning and postconditioning.
Ischemic preconditioning
Many studies in vivo and in vitro have demonstrated that neurons exposed to brief periods of sublethal
anoxia develop resistance to subsequently more prolonged and lethal anoxic insults [16–21]. This phenomenon, known as anoxic preconditioning (APC), was first described in the myocardium [22,23] and only recently in the brain [2,17,24–26]. Consequently, over the last three decades, many efforts have been addressed to identify the molecular mechanisms involved in this phenomenon in order to open up therapeutic avenues for the treatment of cerebral ischemia.
Ischemic preconditioning stimuli
It is generally accepted that preconditioning requires small doses of an otherwise harmful stimulus to induce protection against subsequent injurious challenge [1]. Several distinct preconditioning stimuli can induce tolerance to ischemic brain injury; among them are noninjurious ischemia, cortical spreading depression, brief episode of seizure, exposure to anaesthetic inhalants, and low doses of endotoxin, hyperthermia or heat shock [27,28]. The existence of multiple, diverse preconditioning stimuli able to provide protection against an entirely different type of injury, constitutes the well-known phenomenon of ‘cross-tolerance’ [2]. Accordingly, one stressor can promote cross-tolerance to another, or the same stressors that elicit tolerance in the brain can elicit tolerance in other organs. Many exogenously delivered chemical preconditioning agents, such as inflammatory cytokines and metabolic inhibitors, can also induce ischemic tolerance, raising the possibility that in the future it will be possible to activate pharmacologically these distal pathways in the human brain. Moreover, because inducers and mechanisms of tolerance might have similar features, the induction of tolerance in one organ can, via neural or paracrine mechanisms, spread to other organs. This phenomenon, known as ‘remote preconditioning’ or ‘preconditioning at a distance’, has been recently described both in the brain [29] and in the myocardium [30] after hind limb ischemia or intrarenal occlusion of the aorta, respectively.
Differences in the intensity, duration and ⁄ or frequency of a particular stimulus potentially able to induce protection determine whether that stimulus is too weak to elicit a response, of sufficient intensity to serve as a preconditioning trigger, or too robust to be harmful [1].
In general, it is widely accepted that immediate acquisition of protein-synthesis-independent tolerance is mediated by post-translational modification and that the effective duration is brief. Conversely, there is general agreement that delayed induction of ischemic tolerance requires new protein synthesis and is
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
47 |
Focus on preconditioning and postconditioning G. Pignataro et al.
sustained for a time interval ranging from a few days |
the mitochondria [48] – is able to protect mice against |
|||||||
to a few weeks [1]. In the brain, the time course of |
focal cerebral ischemia [49,50]. In addition, the Ras |
|||||||
ischemic tolerance apparently follows the delayed pat- |
isoform Ki-Ras, which can be activated by NO [46], |
|||||||
tern, suggesting that synthesis of active proteins may |
can post-transcriptionally modulate the mitochondrial |
|||||||
be necessary for full development of ischemic tolerance |
Mn-SOD [51]. Recently, it has been proposed that |
|||||||
[31]. Once induced, the ischemic tolerance is believed |
Mn-SOD may represent the crucial step through which |
|||||||
to last for a few days and to diminished gradually a |
the NO ⁄ Ras ⁄ ERK1 ⁄ 2 pathway promotes neuroprotec- |
|||||||
few weeks after acquisition [32]. |
|
|
tion during preconditioning [52]. |
|
||||
|
|
|
|
However, Mn-SOD is not the only antioxidant |
||||
Mechanisms involved in ischemic |
|
|
enzyme activated during APC. In fact, other groups of |
|||||
|
|
investigators have recently reported |
that glutathione |
|||||
preconditioning neuroprotection |
|
|
||||||
|
|
peroxidase, |
glutathione |
reductase, |
copper ⁄ zinc-SOD |
|||
|
|
|
|
|||||
The molecular mechanisms responsible for the induc- |
and catalase are also elevated during APC [9,45]. |
|||||||
tion and maintenance of ischemic tolerance in the |
|
|
|
|
||||
brain are complex and remain largely undefined. In |
Cellular ionic homeostasis and energy |
|||||||
this context, some studies have demonstrated that sev- |
||||||||
metabolism |
|
|
||||||
eral events, such as the activation of protein kinases |
|
|
||||||
|
|
|
|
|||||
[33,34], the induction of transcription factors [35–37] |
As ischemic preconditioning activates intracellular bio- |
|||||||
and the induction of immediate-early genes [38,39] play |
logical responses prior to a potential lethal insult, it is |
|||||||
pivotal roles in the development of ischemic tolerance. |
expected that an increase of energy metabolism or a |
|||||||
Moreover, oxygen free-radicals, generated during the |
latency in anoxic depolarization after the onset of |
|||||||
preconditioning stimuli, may also elicit ischemic toler- |
ischemic insult might represent the mechanisms by |
|||||||
ance [40] through the neosynthesis of neuroprotective |
which organs strengthen their tolerance when exposed |
|||||||
proteins such as the heat shock protein 72 [41,42], |
to a sublethal insult. In this regard, several experi- |
|||||||
through anti-apoptotic proteins such as B-cell lym- |
ments have been performed, both in vivo and in vitro, |
|||||||
phoma 2 [26,43], and through antioxidant enzymes |
in order to demonstrate that a reduction in energy |
|||||||
[44,45]. Finally, it has also been hypothesized that |
demand and in the activity of ion channels represent |
|||||||
nitric oxide (NO) could bolster intracellular signalling, |
determinant factors for ischemic tolerance [53]. In fact, |
|||||||
which is activated in neurons during APC [46,47]. In |
impairment in voltage-gated potassium channels has |
|||||||
this context, NO acting as an important transducer |
been observed in cortical neurons exposed to brief |
|||||||
would |
activate the Ras ⁄ extracellular |
regulated |
kinase |
noninjurious oxygen and glucose deprivation. Simi- |
||||
(ERK) |
1 ⁄ 2 pathway, thus eliciting |
beneficial |
effects |
larly, in vivo experiments demonstrated that ischemic |
||||
[24,46]. |
|
|
|
preconditioning prevented the inhibition of Na+ ⁄ K+- |
||||
More recently, the inter-relationship among the |
ATPase activity after brain ischemia in hippocampal |
|||||||
mitochondrial function, the preservation of energy |
and cortical neurons of rats exposed to global fore- |
|||||||
metabolism and the appearance of the neuroprotective |
brain ischemia [54]. Regarding calcium homeostasis |
|||||||
effect during APC, has been highlighted in in vitro and |
during ischemic preconditioning, the results of in vivo |
|||||||
in vivo models of cerebral ischemia [29,34]. Indeed, |
experiments in gerbils showed an increase in Ca2+- |
|||||||
ischemic preconditioning has been associated with |
ATPase activity and an enhancement in mitochondrial |
|||||||
improved preservation of energy metabolism and mito- |
calcium sequestration in CA1 hippocampal neurons |
|||||||
chondrial function during the ischemic event [34]. |
after preconditioning [55]. In line with this result, |
|||||||
However, further studies are still needed to define in |
intracellular calcium imaging performed in hippocam- |
|||||||
more detail the molecular mechanisms responsible for |
pal neurons of preconditioned gerbils showed that the |
|||||||
these events. For instance, it is still unclear whether |
increase in [Ca2+]i occurring after anoxic and aglyce- |
|||||||
the protection afforded by APC depends on the reduc- |
mic episodes was markedly inhibited in the ischemic- |
|||||||
tion of energy expenditure, on the increased produc- |
tolerant animals [56]. The molecular mechanisms |
|||||||
tion of high-energy phosphates, on the improvement |
underlying |
this effect |
are still under investigation. |
|||||
of substrate delivery, or, finally, on the improved effi- |
A possible explanation could be increased expression |
|||||||
ciency of energy metabolism. On the other hand, it |
of the Ca2+-ATPase isoform 1 [plasma membrane |
|||||||
has recently been reported that the overexpression |
calcium ATPase 1 (PMCA-1)] as recently demon- |
|||||||
of manganese–superoxide dismutase (Mn-SOD) – a |
strated by Kato et al. [57]. However, the hypothesis |
|||||||
nuclear-encoded mitochondrial enzyme that scavenges |
that a modulation of the expression and activity of the |
|||||||
superoxide generated by the electron-transport chain in |
sodium calcium exchanger (NCX) might play a role in |
|||||||
48 |
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
G. Pignataro et al. |
Focus on preconditioning and postconditioning |
the regulation of calcium and sodium homeostasis during ischemic tolerance cannot be ruled out. It is relevant to mention that NCX gene expression was reduced during cerebral ischemia in rats in a different manner depending on the exchanger isoforms and on the region involved in the insult [58,59].
Also, mitochondria co-operate in the regulation of intracellular Ca2+ homeostasis, both in physiological and in pathological conditions. Although mitochondria have a high capacity for Ca2+ sequestration, excessive amounts of Ca2+, as occurs during ischemia, impair mitochondrial function. This induces a massive uncoupling of oxidative phosphorylation and reduction of mitochondrial membrane potential with consequent mitochondrial permeability transitional pore (MPTP) opening and reversal of the action of ATPase, which in turn hydrolyses, rather than synthesizes, ATP. As a consequence of ATP depletion and increased permeability of the inner mitochondrial membrane, cell death occurs. Studies performed both in the heart and in the brain suggested that the inhibition of MPTP opening and its signalling cascade represent crucial events responsible for cytoprotection observed in ischemic preconditioning [60,61]. The molecular mechanisms underlining these effects are still the objects of investigation. Nitrite and protein kinases have been proposed as possible MPTP regulators [62,63].
Nitric oxide
Both in the heart and in the brain, NO emerged as an important mediator of tolerance. In fact, it has been reported that inducible NO synthase plays a crucial role in delayed preconditioning and tolerance in the heart but not in the brain [64,65]. Endothelial NO synthase (eNOS) plays a controversial role in the early phase of cardiac preconditioning in adult heart, whereas it appears to be primarily involved in ischemic preconditioning of immature brain [66]. One potential mechanism for eNOS-induced neuroprotection would be vasodilation of cerebral vessels supplying the ischemic penumbra with augmentation of blood flow. Other potential mechanisms might involve leukocyte–endo- thelial interactions, or platelet–endothelial interactions modulated by eNOS. However, eNOS might not play a prominent role in adult brain. By contrast, neuronally derived NO synthase (nNOS) seems to play a relevant role in neuroprotection induced by ischemic preconditioning in mature neurons [46]. This protection requires N-methyl-d-aspartate receptor activation, calcium influx and new protein synthesis. In fact, nNOS-derived NO triggers p21Ras activation, which, in turn, stimulates downstream Ras-dependent signal-
ling and intracellular pathways, leading to changes in gene expression, stimulation of programs related to neuronal growth and promotion of neuronal survival. Recent findings demonstrated that the increase in NO production observed during preconditioning might affect mitochondrial function, either modulating MPTP opening [62] or increasing the expression and activity of mitochondrial Mn-SOD [52], thus regulating ROS production. Although different sources of NO might activate ischemic preconditioning, common final mediators might exist. It has been recently hypothesized that NO interacts with at least two signalling
pathways |
important |
for |
neuronal |
survival: the |
||
Ras ⁄ Raf ⁄ mitogen-activated |
protein |
kinase |
kinase ⁄ |
|||
ERK cascade [46], |
and |
the |
phosphatidylinositol |
|||
3-kinase |
(PI3K) ⁄ Akt |
pathway |
[67]. These |
pathways |
||
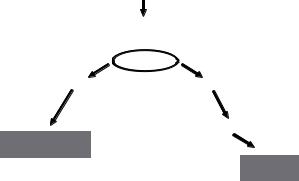
may represent the unifying mechanisms that underlie protection (Fig. 1).
Kinases
The early phase of ischemic preconditioning is characterized by rapid post-translational modification of pre-existing proteins through signalling pathways that involve protein kinase C (PKC) [68] and mitogen-acti- vated protein kinase (MAPK). The late preconditioning is mediated by protective gene expression and by the synthesis of new protective proteins. This mechanism involves redox-sensitive activation of
Preconditioning
nNOS
 NO RAS
NO RAS
ERK |
PI3K |
Akt
Mn-SOD
Neuroprotection ?
Fig. 1. Hypothetical neuroprotective mechanisms mediated by ischemic preconditioning NO interacts with two signalling pathways important to neuronal survival: the Ras ⁄ ERK pathway, and the PI3K ⁄ Akt pathway. A preconditioning stimulus activates nNOS and induces NO production. NO activates the G-protein Ras, which in turn phosphorylates and activates ERK. ERK post-transcriptionally modulates the expression and activity of the mitochondrial Mn-SOD, thus inducing neuroprotection. Moreover, activated Ras also stimulates the PI3K ⁄ Akt pathway, which in turn activates proteins involved in preconditioning-induced neuroprotection.
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
49 |
Focus on preconditioning and postconditioning |
G. Pignataro et al. |
transcriptional factors through PKC and tyrosine kinase signalling pathways that are in common with the early phase of preconditioning.
Important steps in the signalling pathway of ischemic preconditioning are represented by the activation of phospholipase D, tyrosine kinase and MAPK, with interactions among these pathways not yet entirely elucidated [69]. Oxidative stress has been proposed as a possible activator of these enzymes in various cell types, [70] suggesting that these molecular mechanisms might be responsible for ROS-induced ischemic tolerance, independently from PKC activation. On the other hand, ROS mediates transactivation of receptor tyrosine kinase through Src, a family of tyrosine kinases known to be activated by ROS and to interact with many signalling proteins including PKC and PI3K [71]. Such protein–protein interaction forms a signalling module that may be important in integrating protective signal transduction in ischemic preconditioning [72].
MAPKs, a serine ⁄ threonine protein kinase family, play a crucial role in triggering the intracellular events leading to the activation of the adaptive response observed in ischemic preconditioning, both in the heart [73] and in the brain [11,74]. Interestingly, post-ischemic activation of Akt ⁄ protein kinase B may contribute to the induction of ischemic tolerance [75]. In fact, in the above-mentioned papers, it has been reported that Akt was activated after sublethal ischemia, and inhibition of Akt activity resulted in attenuation of ischemic tolerance.
Ischemic postconditioning
Ischemic postconditioning stimuli
Unlike ischemic preconditioning, the neuroprotective strategy named ischemic postconditioning is a relatively novel concept [10,76]. Rapid revascularization of the occluded vessels and timely reperfusion is one of the most effective approaches currently used for acute ischemic stroke. However, it has been repeatedly demonstrated that during the early reperfusion phase reactive oxygen species are generated and intracellular free Ca2+ overload may occur, potentially leading to additional injury [77]. In an attempt to attenuate the injurious early hyperemic response after reperfusion, a novel neuroprotective procedure termed ischemic ’postconditioning’ has been reported. This neuroprotective strategy is defined as a repetitive series of brief interruptions of reperfusion applied immediately after ischemia. Repeated cycles of brief reperfusion and re-occlusion were initially demonstrated to reduce the
infarct size after cardiac ischemia, both in the experimental setting [78] and in the clinical setting [79]. More recently, ischemic postconditioning has also been shown to attenuate neuronal damage in rodent models of spinal cord [80], and focal [10,11,76] and global [8,81] ischemic injury.
In particular, brain neuroprotection induced by postconditioning has been achieved by subjecting the brain to different cycles of short, nondangerous ischemia applied after harmful ischemia. In focal ischemia, two models have been used to date to induce postconditioning: in the first model, permanent distal occlusion of the middle cerebral artery was followed by a series of occlusion of both common carotid arteries (CCAs) [10]; in the second model, harmful transient middle cerebral artery occlusion was followed by a series of brief noninjurious middle cerebral artery occlusions and reperfusions [11,76]. Ischemic postconditioning was achieved also in animals subjected to global ischemia induced by occlusion of the CCAs and of the two vertebral arteries, 4-vessel occlusion, by subjecting the animals to different cycles of noninjurious CCA occlusion.
Recently, this neuroprotective mechanism has been reproduced also in in vitro preparations (i.e. in hippocampal organotypic slice cultures and primary neurons subjected to harmful oxygen glucose deprivation), followed by noninjurious cycles of oxygen glucose deprivation and re-oxygenation [11,76,82].
Interestingly, the cross-tolerance phenomenon, in which one stressor induces protection against a different stressor, occurs even in postconditioning. In fact, it has been shown that not only brief periods of ischemia produced by repeated interruptions of reperfusion, but also other pharmacological strategies that have been previously used as preconditioning stimuli, can be used for postconditioning [8]. Collectively, it is possible to state that several treatments can be used as postconditioning stimuli and are effective for ischemic tolerance induction (i.e. short ischemia, hypoxia, isoflurane, norepinephrine and 3-nitropropionic acid) (Fig. 2).
Mechanisms involved in ischemic postconditioning neuroprotection
Little is known about the postconditioning protective mechanisms against cerebral ischemia. However, because postconditioning, by definition, is performed after the insult, it is essential to characterize this phenomenon in greater detail, in the hope of developing an effective clinical approach to treat stroke. It has been hypothesized that ischemic postconditioning reduces infarct size after focal stroke as a function of
50 |
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
G. Pignataro et al. |
Focus on preconditioning and postconditioning |
|
|
Blood vessel occlusion |
|
|
|
|
|
|
|
Chemical treatment |
|
|
|
|
(i.e. 3-Nitropropionic |
|
|
|
|
acid, Norepinephrine, |
|
|
|
|
|
|
Temperature (hypothermia) |
||
Isoflurane) |
|
|
||
|
|
|
|
|
Post conditioning
Others? Hormones
(i.e. urocortin)
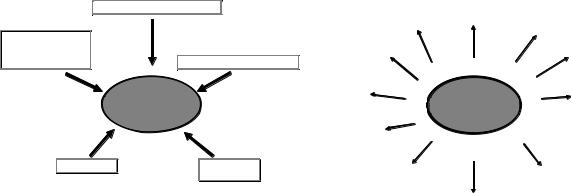
Fig. 2. Stimuli able to induce cerebral ischemic postconditioning. Neuroprotection induced by ischemic postconditioning can be induced by applying a myriad of different stimuli after harmful ischemia. Within them are: hypothermia, blood vessel occlusion, hormone administration and treatment with chemical compounds like 3-nitropropionic acid, norepinephrine and isofluorane.
stroke severity, probably by reducing apoptosis and free radical products [83]. In fact, the expression and the activity of endogenous antioxidant enzymes such as Mn-SOD are increased in the brain of animals subjected to postconditioning treatment, thus playing an important neuroprotective role in this neuroprotective strategy [84]. Additionally, it has been shown that postconditioning enhances ERK1 ⁄ 2 expression and promotes an increase in phosphorylated Akt expression and activity [11,85]. In particular, the role of the Akt pathway in postconditioning has probably received major attention in different animal models of brain ischemic postconditioning [11,85,86]. In summary, although several groups have reported that postconditioning reduces ischemic damage in the brain, many outstanding issues remain elusive, such as the number of ischemia ⁄ reperfusion cycles as well as the duration of reperfusion and occlusion for postconditioning, therapeutic time-windows and, more importantly, the underlying protective mechanisms of postconditioning. Future studies should address these and other missing points in an attempt to clarify in greater detail this important neuroprotective strategy. The mechanisms identified to date as responsible for postconditioning (Fig. 3) neuroprotection will be now briefly reviewed below.
Cellular ionic homeostasis and energy metabolism
Given the potentially lethal consequences of intracellular Ca2+ overload [87], it is relevant to examine whether Ca2+ homeostasis is altered during postconditioning neuroprotection.
Activation of cyclooxygenase and
prostacyclin
Suppressing ER stress
Activation of apoptosis via inhibition of JNK and p38
Signalosome |
|
NO |
|
|
|
ROS |
Post |
Sphingosine |
|
||
|
|
|
|
conditioning |
|
RISK pathway
 mGlu1/mGlu5-
mGlu1/mGlu5-
PI3K-Akt
Activation of A1 receptors and Activation of opioid receptors K+(ATP) channels
Reduced oxidative stress
Fig. 3. Receptors and proteins involved in the neuroprotection elicited by cerebral ischemic postconditioning. Postconditioning exerts its neuroprotective action by acting on different proteins involved in ionic transport across the plasma membrane, G-protein-coupled membrane receptors, ionic receptors and kinase pathways. A1 receptor, adenosine receptor 1; ER, endothelium receptor; JNK, c-jun N-terminal kinase; mGLU1, metabotropic glutamate receptor 1; mGLU5, metabotropic glutamate receptor 5; NO, nitric oxide; PI3K, phosphatidylinositol 3-kinase; RISK, reperfusion injury salvage kinase; ROS, reactive oxygen species.
Since the early 1970s, the role of proteins involved in Ca2+ homeostasis during cerebral ischemia have been studied in terms of expression, activity and pharmacological relevance. Na+ ⁄ Ca2+ exchangers and plasma-membrane Ca2+ pumps are crucial for intracellular Ca2+ homeostasis and Ca2+ signalling. Different neurotoxic stimuli are able to modify the expression and the function of these two families of transporters. In fact, 2–3 h of exposure to 300 lm H2O2 induces a significant downregulation of all NCXs and PMCAs at the RNA and protein levels [88]. In addition, in previous work it has been shown that NCX gene expression after permanent middle cerebral artery occlusion in rats is regulated in a differential manner, depending on the exchanger isoform (NCX1, )2, or )3) and the region involved in the insult (i.e. ischemic core, periinfarct areas or spared regions) [58,59].
Na+ ⁄ H+ exchange and Na+ ⁄ K+ ⁄ 2Cl) cotransport are also critical ion transporters as they contribute to the regulation of intracellular pH and cell volume [89]. This dual role, however, makes it difficult to predict whether a change in the expression and in the activity of these transporters may promote or impair the survival of cells to ischemia; for example, enhanced Na+ ⁄ H+ exchange would help to reduce the intracellular pH but promote cell swelling and further Ca2+ influx through Na+ ⁄ Ca2+ exchange. Experiments carried out in cortical astrocyte cultures from Na+ ⁄ H+ exchanger isoform 1-deficient [NHE1() ⁄ ))] mice
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
51 |
Focus on preconditioning and postconditioning |
G. Pignataro et al. |
suggested that NHE1 deficiency actually attenuated damage induced by conditions mimicking cerebral ischemia in vitro [90], and this notion was confirmed in vivo [91]. Similar data were obtained with genetic manipulations of the Na+ ⁄ K+2Cl) cotransporter isoform 1 (NKCC1) in mice and by the inhibition of NKCC1 using bumetanide [91]. Another important player able to modulate intracellular Ca2+ homeostasis during ischemia is the pH-sensitive channel ASIC1a, a channel belonging to the family of acid-sensing ionic channels and widely expressed within the central nervous system [92]. The important role played by this channel in the development of ischemic damage is testified by the fact that its pharmacological blockade or its genetic ablation induced a dramatic reduction of the consequences of stroke mimicked in mice and rats occluding the middle cerebral artery [93,94].
According to these data, one could expect ischemic neuroprotection induced by postconditioning to be associated with the downregulation of NHE1, ASIC1a and ⁄ or NKCC1, and the upregulation of NCX and PMCA.
Although the results obtained in models of cerebral ischemia are encouraging, several other issues need to be addressed in order to clarify in greater detail the role of ionic homeostasis in the course of ischemic postconditioning.
NO
The role of NO in ischemic postconditioning has not yet been studied. However, it is possible to hypothesize that it plays a fundamental role in the neuroprotective mechanisms induced by postconditioning. The first reason why this represents a plausible hypothesis resides in the fact that NO is an important mediator of the preconditioning neuroprotection, and mechanisms responsible for postconditioning protection appear to be similar to those identified in conventional preconditioning [95]. Second, it has been reported that NO represents an important mediator of postconditioning neuroprotection exerted in organs different from the brain (i.e. the heart and the kidney). Therefore, NO appears to be a reliable candidate in mediating postconditioning neuroprotection.
Kinases
In cardiac postconditioning, the signalling pathways involved in postconditioning neuroprotection include the activation of prosurvival protein kinases, and these same kinases may also be effectors of cardiac preconditioning [6]. Recent studies suggest that postcondi-
tioning in the brain may also involve the activation of the family of protein kinases called reperfusion injury salvage kinases [11,76,85]. Among them, Akt and ERK have been shown to regulate ischemic precondi- tioning-induced neuroprotection, both in the heart and in the brain [26,74]. Interestingly, during harmful ischemia, Akt is transiently phosphorylated and consequently activated, only for a short interval of time after reperfusion, whereas after postconditioning, the phosphorylation of Akt persists for longer, still being present in the phosphorylated form even 24 h later. ETK and P38 MAPK also show similar activation profiles after postconditioning.
However, while the role played by phosphorylated AKT in mediating postconditioning neuroprotection has been sufficiently elucidated in different models of cerebral postconditioning [11,85], contrasting results have been obtained with phosphorylated ERK1 ⁄ 2 and phosphorylated c-jun N-terminal kinase (JNK) in postconditioning. In fact, while the expression of phosphorylated ERK and phosphorylated JNK is increased after postconditioning, their inhibition is able to revert the neuroprotection induced by postconditioning only under some experimental conditions [85]. These data support distinct cell-signalling mechanisms of preconditioning versus postconditioning in the brain and suggest a role for AKT and its downstream signalling cascade in mediating postconditioning-induced neuroprotection.
The pharmacological postconditioning effect of the activation of AKT after harmful ischemia may account for the delayed activation of cell-death pathways, because inhibition of AKT has been shown to speed cell death after ischemia [50]. Specific targets of AKT have not been shown in postconditioning; potential effectors include (a) cellular responsive element-binding protein and (b) B-cell lymphoma 2 upregulation [96] (both of which are involved in preconditioning neuroprotection [26,97]), (c) activation of the Akt ⁄ glycogen synthase kinase 3 beta signalling pathway [98] or (d) the prosurvival effect on Bad and caspase 9 [99,100]. In a recent publication, the role of AKT has been confirmed also in in vitro experiments [82]. In fact, it has been shown that activation of the metabotropic glutamate receptor 1 ⁄ metabotropic glutamate receptor 5 ⁄ PI3K ⁄ Akt signalling pathway plays a crucial role in the mechanisms of postconditioning induced in hippocampal organotypic slice cultures [82].
From these results, postconditioning stimulus results in the prolonged activation of AKT, which has also been shown to mediate intrinsic protection to ischemia in models of ischemic preconditioning. In addition, recent studies showed that postconditioning protection
52 |
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
G. Pignataro et al. |
Focus on preconditioning and postconditioning |
is correlated with the inhibition of ERK1 ⁄ 2 and JNK activities, promotion of ePKC phosphorylation and reduction of dPKC cleavage.
Inflammation mediators
Inflammation has been indicated as an important process occurring during cerebral ischemic injury [101– 103]. In fact, after cerebral ischemic injury, macrophages, endothelial cells, astrocytes, fibroblasts and neurons produce cytokines to fuel inflammatory reactions. Among these cytokines, interleukin (IL)-1b and tumor necrosis factor-alfa (TNF-a) play a determinant role [104–106]. In addition to attracting leukocytes into ischemic regions and promoting tissue necrosis, IL-1b and TNF-a stimulate intercellular adhesion molecule-1 (ICAM-1) expression [107], which is critical in leukocyte accumulation and infiltration into the site of injury [108]. The upregulation of these inflammatory mediators during cerebral ischemia promotes the adherence and infiltration of recruited blood-borne leukocytes during reperfusion. The leukocytes exacerbate brain injury by obstructing capillaries and reducing blood flow, and by releasing cytotoxic products [109]. Consequently, these inflammatory mediators and the infiltration of leukocytes contribute to irreversible infarction [103]. Several studies showed that the accumulation of leukocytes was attenuated by ischemic postconditioning in myocardial ischemic injury [14,78]. Recently, it has been reported that increased levels of markers of inflammation, myeloperoxidase activity and expression of IL-1b, TNF-a and ICAM-1, were reduced by ischemic postconditioning treatment. Therefore, ischemic postconditioning ameliorates ischemic injury by reducing inflammatory responses after focal cerebral ischemia. Several studies suggest that the mechanism by which postconditioning would reduce the inflammation cascade seems to be ascribed to the suppression of oxygen radical overproduction [10,78].
Future perspectives
Studies on postconditioning and preconditioning as neuroprotective strategies are currently being published at what appears to be a near-exponential rate. The burst of studies on this issue should help to advance our understanding of their mechanistic basis and, in turn, their clinical potential. An important point to be underlined is that the endogenous survival mechanisms activated in response to postconditioning and preconditioning do not depend on differences in drug pharmacokinetics or administration protocols that can confound the translation of neuroprotective strategies
from rodents to humans. Therefore, the identification of intrinsic cell-survival pathways should provide more direct opportunities for translational neuroprotection trials. Although primarily focused on stroke at present, we might find that the innate regulatory schemes that underlie postconditioning and preconditioning to ischemia are also applicable to protecting the brain from other acute and chronic neurodegenerative disorders.
Acknowledgements
The present study was supported by grants from contribution of ‘Ministero Affari Esteri, Direzione Generale per la Promozione e la Cooperazione Culturale Fondi Italia-Cina Legge 401 ⁄ 1990 2007–2008. Ricerca Finalizzata RF-FSL-2006 352059. COFIN 2006.
References
1 Dirnagl U, Simon RP & Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26, 248–254.
2Kirino T (2002) Ischemic tolerance. J Cereb Blood Flow Metab 22, 1283–1296.
3Gidday JM (2006) Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 7, 437–448.
4Gladstone DJ, Black SE & Hakim AM (2002) Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke 33,
2123–2136.
5Hausenloy DJ, Tsang A, Mocanu MM & Yellon DM (2005) Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart
Circ Physiol 288, H971–H976.
6Hausenloy DJ, Tsang A & Yellon DM (2005) The reperfusion injury salvage kinase pathway: a common
target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med 15, 69–75.
7Yellon DM & Hausenloy DJ (2005) Realizing the clinical potential of ischemic preconditioning and
postconditioning. Nat Clin Pract Cardiovasc Med 2, 568–575.
8Burda J, Danielisova V, Nemethova M, Gottlieb M, Matiasova M, Domorakova I, Mechirova E, Ferikova M, Salinas M & Burda R (2006) Delayed postconditionig initiates additive mechanism necessary for survival of selectively vulnerable neurons after transient ische-
mia in rat brain. Cell Mol Neurobiol 26, 1141–1151.
9Danielisova V, Nemethova M, Gottlieb M & Burda J (2006) The changes in endogenous antioxidant enzyme activity after postconditioning. Cell Mol Neurobiol 26, 1181–1191.
10Zhao H, Sapolsky RM & Steinberg GK (2006) Interrupting reperfusion as a stroke therapy: ischemic
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
53 |
Focus on preconditioning and postconditioning |
G. Pignataro et al. |
postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab 26, 1114–1121.
11Pignataro G, Meller R, Inoue K et al. (2008) In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab 28, 232–241.
12Burda J, Marsala M, Radonak J & Marsala J (1991) Graded postischemic reoxygenation ameliorates inhibition of cerebral cortical protein synthesis in dogs.
J Cereb Blood Flow Metab 11, 1001–1005.
13Burda J, Gottlieb M, Vanicky I, Chavko M & Marsala J (1995) Short-term postischemic hypoperfusion improves recovery of protein synthesis in the rat brain cortex. Mol Chem Neuropathol 25, 189–198.
14Kin H, Zatta AJ, Lofye MT et al. (2005) Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 67, 124–133.
15Allen BS, Halldorsson AO, Barth MJ & Ilbawi MN (2000) Modification of the subclavian patch aortoplasty for repair of aortic coarctation in neonates and infants. Ann Thorac Surg 69, 877–880; discussion 881.
16Kitagawa K, Matsumoto M, Tagaya M et al. (1990) ‘Ischemic tolerance’ phenomenon found in the brain.
Brain Res 528, 21–24.
17Kitagawa K, Matsumoto M, Kuwabara K et al. (1991) ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res 561, 203–211.
18Glazier SS, O’Rourke DM, Graham DI & Welsh FA (1994) Induction of ischemic tolerance following brief focal ischemia in rat brain. J Cereb Blood Flow Metab 14, 545–553.
19Miyashita K, Abe H, Nakajima T et al. (1994) Induction of ischaemic tolerance in gerbil hippocampus by pretreatment with focal ischaemia. Neuroreport 6, 46–48.
20Simon RP, Niiro M & Gwinn R (1993) Prior ischemic stress protects against experimental stroke. Neurosci Lett 163, 135–137.
21Gidday JM, Fitzgibbons JC, Shah AR & Park TS (1994) Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett 168, 221–224.
22Murry CE, Jennings RB & Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136.
23Meldrum DR, Cleveland JC Jr, Rowland RT, Banerjee A, Harken AH & Meng X (1997) Early and delayed preconditioning: differential mechanisms and additive protection. Am J Physiol 273, H725–H733.
24Dawson VL & Dawson TM (2000) Neuronal ischaemic preconditioning. Trends Pharmacol Sci 21, 423–424.
25Schaller B & Graf R (2002) Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 249, 1503–1511.
26Meller R, Minami M, Cameron JA et al. (2005) CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab 25, 234– 246.
27Kobayashi S, Harris VA & Welsh FA (1995) Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab 15, 721–727.
28Plamondon H, Blondeau N, Heurteaux C & Lazdunski M (1999) Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and K(ATP) channels. J Cereb Blood Flow Metab 19, 1296–1308.
29Dave KR, Saul I, Prado R, Busto R & Perez-Pinzon MA (2006) Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci Lett 404, 170– 175.
30Weinbrenner C, Schulze F, Sarvary L & Strasser RH (2004) Remote preconditioning by infrarenal aortic occlusion is operative via delta1-opioid receptors and free radicals in vivo in the rat heart. Cardiovasc Res 61, 591–599.
31Barone FC, White RF, Spera PA et al. (1998) Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 29, 1937–1950; discussion 1950–1951.
32Kirino T, Tsujita Y & Tamura A (1991) Induced tolerance to ischemia in gerbil hippocampal neurons.
J Cereb Blood Flow Metab 11, 299–307.
33Lange-Asschenfeldt C, Raval AP, Dave KR, MochlyRosen D, Sick TJ & Perez-Pinzon MA (2004) Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab 24, 636–645.
34Perez-Pinzon MA, Dave KR & Raval AP (2005) Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxid Redox Signal 7, 1150–1157.
35Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J & Hallenbeck JM (2002) TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kap- paB transactivation: the role of NF-kappaB association with p300 adaptor. J Cereb Blood Flow Metab 22, 142– 152.
36Jones NM & Bergeron M (2001) Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab 21, 1105–1114.
37Digicaylioglu M & Lipton SA (2001) Erythropoietinmediated neuroprotection involves cross-talk between
54 |
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
G. Pignataro et al. |
Focus on preconditioning and postconditioning |
Jak2 and NF-kappaB signalling cascades. Nature 412, 641–647.
38Rybnikova E, Vataeva L, Tyulkova E et al. (2005) Mild hypoxia preconditioning prevents impairment of passive avoidance learning and suppression of brain NGFI-A expression induced by severe hypoxia. Behav Brain Res 160, 107–114.
39Patel A, Van de Poll MC, Greve JW et al. (2004) Early stress protein gene expression in a human model of ischemic preconditioning. Transplantation 78, 1479– 1487.
40Ravati A, Ahlemeyer B, Becker A & Krieglstein J (2000) Preconditioning-induced neuroprotection is mediated by reactive oxygen species. Brain Res 866, 23–32.
41Massa SM, Swanson RA & Sharp FR (1996) The stress gene response in brain. Cerebrovasc Brain Metab Rev 8, 95–158.
42Rejdak R, Rejdak K, Sieklucka-Dziuba M, Stelmasiak Z & Grieb P (2001) Brain tolerance and preconditioning. Pol J Pharmacol 53, 73–79.
43Brambrink AM, Schneider A, Noga H et al. (2000) Tolerance-Inducing dose of 3-nitropropionic acid modulates bcl-2 and bax balance in the rat brain: a potential mechanism of chemical preconditioning. J Cereb Blood Flow Metab 20, 1425–1436.
44Toyoda T, Kassell NF & Lee KS (1997) Induction of ischemic tolerance and antioxidant activity by brief focal ischemia. Neuroreport 8, 847–851.
45Arthur JW & Wilkins MR (2004) Using proteomics to mine genome sequences. J Proteome Res 3, 393–402.
46Gonzalez-Zulueta M, Feldman AB, Klesse LJ et al. (2000) Requirement for nitric oxide activation of p21(ras) ⁄ extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA 97, 436–441.
47Huang PL (2004) Nitric oxide and cerebral ischemic preconditioning. Cell Calcium 36, 323–329.
48Hirai K, Hayashi T, Chan PH et al. (2004) PI3K inhibition in neonatal rat brain slices during and after hypoxia reduces phospho-Akt and increases cytosolic cytochrome c and apoptosis. Brain Res Mol Brain Res 124, 51–61.
49Murakami K, Kondo T, Kawase M et al. (1998) Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci 18, 205–213.
50Noshita N, Sugawara T, Fujimura M, Morita-Fujim- ura Y & Chan PH (2001) Manganese superoxide dismutase affects cytochrome c release and caspase-9 activation after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21, 557–567.
51Santillo M, Mondola P, Seru R et al. (2001) Opposing functions of Kiand Ha-Ras genes in the regulation of redox signals. Curr Biol 11, 614–619.
52Scorziello A, Santillo M, Adornetto A et al. (2007) NO-induced neuroprotection in ischemic preconditioning stimulates mitochondrial Mn-SOD activity and expression via Ras ⁄ ERK1 ⁄ 2 pathway. J Neurochem 103, 1472–1480.
53Stenzel-Poore MP, Stevens SL, Xiong Z et al. (2003) Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362, 1028–1037.
54de Souza Wyse AT, Streck EL, Worm P, Wajner A, Ritter F & Netto CA (2000) Preconditioning prevents the inhibition of Na+,K+-ATPase activity after brain ischemia. Neurochem Res 25, 971–975.
55Ohta S, Furuta S, Matsubara I, Kohno K, Kumon Y & Sakaki S (1996) Calcium movement in ischemia-tol- erant hippocampal CA1 neurons after transient forebrain ischemia in gerbils. J Cereb Blood Flow Metab 16, 915–922.
56Shimazaki K, Nakamura T, Nakamura K et al. (1998) Reduced calcium elevation in hippocampal CA1 neurons of ischemia-tolerant gerbils. Neuroreport 9, 1875–1878.
57Kato K, Shimazaki K, Kamiya T et al. (2005) Differential effects of sublethal ischemia and chemical preconditioning with 3-nitropropionic acid on protein expression in gerbil hippocampus. Life Sci 77, 2867– 2878.
58Pignataro G, Gala R, Cuomo O et al. (2004) Two sodium ⁄ calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke 35, 2566–2570.
59Boscia F, Gala R, Pignataro G et al. (2006) Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+ ⁄ Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J Cereb Blood Flow Metab 26, 502–517.
60Halestrap AP, Clarke SJ & Khaliulin I (2007) The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta 1767, 1007–1031.
61Dirnagl U & Meisel A (2008) Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology 55, 334–344.
62Shiva S, Sack MN, Greer JJ et al. (2007) Nitrite augments tolerance to ischemia ⁄ reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204, 2089–2102.
63Zhao H, Sapolsky RM & Steinberg GK (2006) Phos- phoinositide-3-kinase ⁄ akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol 34, 249–270.
FEBS Journal 276 (2009) 46–57 Journal compilation ª 2008 FEBS. No claim to original Italian government works |
55 |