

Увеличение осмотического давления (гиперосмия): а) результат катаболических процессов — расщепление крупных молекул на более мелкие; б) увеличение концентрации ионов К+, Са2+ (гипериония); в) дисиония — изменение соотношения между отдельными ионами, К+ высвобождается из гибнущих клеток.
Последствия: гипергидратация очага воспаления, стимуляция эмиграции лейкоцитов, изменение тонуса стенок сосудов, формирование чувства боли.
Гиперонкия — увеличение концентрации белка в очаге воспаления, альбумины выходят в ткань вместе с экссудатом. Это ведет к развитию отека.
Изменение поверхностного заряда клеток (как правило, снижение). Обусловлено нарушением водно-электролитного баланса в воспаленной ткани.
♦Механизмы развития: нарушение энергообеспечения трансмембранного переноса ионов и развитие электролитного дисбаланса.
♦Последствия: изменение порога возбудимости клеток, потенцирование миграции фагоцитов за счёт электрокинеза; стимуляция кооперации клеток в связи со снижением величины отрицательного поверхностного их заряда, нейтрализацией его или даже перезарядкой.
Изменения коллоидного состояния межклеточного вещества и гиалоплазмы клеток в очаге воспаления.
♦ Механизмы развития:
- Ферментативный и неферментативный гидролиз макромолекул (гликозаминогликанов, белков, протеогликанов).
-Фазовые изменения микрофиламентов, облегчающие переход их состояния из геля в золь и наоборот.
+♦ Последствия (основное): увеличение тканевой проницаемости.
Уменьшение поверхностного натяжения клеточных мембран. Обусловлено изменениями структуры молекул плазмолеммы.
♦Механизмы развития: воздействие на клеточные мембраны значительного количества поверхностноактивных веществ (фосфолипидов, ВЖК, К+, Са2+).
♦Последствия: облегчение подвижности клетки и потенцирование адгезии клеток при фагоцитозе.
22.Воспаление. Сосудистые реакции при воспалении. Экссудация, механизмы развития, роль медиаторов. Значение экссудации.
Экссудация — компонент воспаления, включающий в себя сосудистые и клеточные реакции.
Этот компонент воспаления включает в себя:
•сосудистые реакции и изменения кровообращения в очаге воспаления;
•выход жидкой части крови из сосудов (экстравазация);
•выход лейкоцитов в очаг воспаления (эмиграция лейкоцитов);
•развитие фагоцитоза.
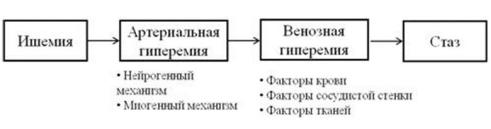
Спазм возникает рефлекторно в ответ на действие флогогена.
Атрериальная гиперемия — результат действия медиаторов воспаления, снижения рН и увеличения осмолярности ткани в очаге повреждения, что ведет к увеличению притока артериальной крови, увеличению скорости кровотока, увеличению гидростатического давления в капиллярах и открытию ранее не функционировавших сосудов.
Венозная гиперемия — результат сдавления венозных сосудов жидкостью, вышедшей в ткань (экссудатом), это ведет к микротромбозу вен и лимфатических сосудов, набуханию эндотелия, краевому стоянию лейкоцитов, сладжированию крови, развитию толчкообразного и маятникообразного кровотока, и в итоге развивается венозный стаз — полная остановка кровотока. Формируется барьер, ограничение очага воспаления.
Непосредственно экссудация — выход жидкой части крови, электролитов, белков и форменных элементов крови из сосудов в очаг воспаления. Жидкость, которая выходит из сосудов (экссудат) формирует отек.
Выход клеток в очаг воспаления — эмиграция. Развитию эмиграции способствуют следующие условия:
–увеличение проницаемости сосудистой стенки;
–выход белка в ткань (гиперонкия);
–увеличение осмотического давления в очаге повреждения;
–увеличение гидростатического давления внутри сосудов.
С момента венозной гиперемии создаются благоприятные условия для выхода клеток во внесосудистое пространство и развития клеточных реакций в очаге воспаления. Замедление скорости кровотока при венозной гиперемии ведет к развитию феномена краевого стояния лейкоцитов (маргинация) и прилипанию их к эндотелию (адгезия), через 2–4 часа сосудистая стенка покрывается слоем лейкоцитов и приобретает вид «булыжной мостовой», а еще через 1–3 часа начинается массивный выход лейкоцитов в ткань (эмиграция).
Место выхода — посткапиллярные венулы.
Медиаторы воспаления и цитокины (С5а, ЛТ В4, ФАТ, ИЛ-1, ИЛ-8, γ-интерферон, ФНО-α) воздействуют на клетки эндотелия, вызывая экспрессию на мембране этих клеток особых молекул адгезии и начинается процесс взаимодействия эндотелиальных клеток с лейкоцитами. Молекулы адгезии — лиганд-рецепторы для межклеточных взамодействий.
1. Селектины — лектиновые молекулы, трансмембранные белки, концевой домен этих белков способен связывать олигосахариды (т.е. белок-лектин). Селектины опосредуют раннюю стадию маргинации — обратимую адгезию клеток.
2.Интегрины — димерные трансмембранные белки, отвечают за поздние стадии адгезии лейкоцитов и тромбоцитов к эндотелию и частично за диапедез клеток, контактные взаимодействия лейкоцитов и тромбоцитов в очаге воспаления.
3.Семейство иммуноглобулинов — являются трансмембранными
белками с пятью доменами, экспрессируются преимущественно эндотелием после активации цитокинами (ИЛ-1, ИЛ-8, γ-интерферон, ФНО-α), они обеспечивают отсроченную адгезию, прохождение лейкоцитов через сосудистую стенку.
4. Адресины — белки клеток внутренней выстилки высокоэндотелиальных венул (сосуды лимфоидных органов), они играют роль при развитии хронического воспаления, когда происходит усиленный ангиогенез и формируются окна усиленной миграции лейкоцитов (например, в суставы при аутоиммунном воспалении).
Хемоаттрактанты (липополисахариды бактериальной стенки Гр (-), ЛТ В4, ФАТ, ИЛ-8, С5а) — вызывают хемотаксис, активное перемещение лейкоцитов в направлении наибольшей концентрации этих веществ. Последовательность выхода лейкоцитов в очаг воспаления — через 2 часа (максимум через 4–6 часов) — нейтрофилы, моноциты — через 16–24 часа.
Значение
Смысл выхода лейкоцитов в очаг воспаления — осуществление фагоцитоза (поглощение и нейтрализация чужеродного агента).
23. Сравнительная патология воспаления (И.И. Мечников). Эмиграция лейкоцитов (L) в очаг воспаления. Фагоцитоз.
Классическое описание сравнительной патологии воспаления дал И. И. Мечников, показав, что
воспаление всегда представляет собой активную реакцию организма, на какой бы ступени эволюционного развития он ни находился.
Воспаление встречается в различных формах у всех представителей животного мира. Усложнение организации животного сопровождается усложнением воспалительной реакции. Как и другие патологические процессы, воспаление эволюционирует с эволюцией животных видов. У животных, лишенных кровеносных сосудов (губки, кишечнополостные, иглокожие), воспаление выражается в скоплении амебоидных соединительнотканных клеток (амебоциты) вокруг места повреждения. И. И. Мечников вводил шип розы в прозрачный колокол медузы и наблюдал скопление амебоцитов вокруг поврежденного участка ткани. Эта реакция и представляла собой воспаление.
У высших беспозвоночных (ракообразные, насекомые), имеющих кровеносную систему открытого типа, воспаление также выражается в скоплении кровяных клеток - лимфогематоцитов - в месте повреждения. Изменения кровообращения в воспаленной ткани, характерные для позвоночных животных и человека, у беспозвоночных не возникают.
Развитие кровеносной системы и ее нервной регуляции у позвоночных животных и у человека значительно усложнило воспалительную реакцию. Расстройства кровообращения в воспаленной ткани являются важнейшими выражениями воспаления. Кроме того, существенное значение в развитии воспаления приобрела нервная система. Участие кровяных клеток в воспалении у высших животных и у человека проявляется выходом лейкоцитов в воспаленную ткань. Кроме того, наблюдается размножение местных соединительнотканных клеток (гистиоцитов, фибробластов) в очаге воспаленной ткани.
Эмиграция лейкоцитов в очаг воспаления – в фазу ВГ; место выхода – посткапиллярные венулы Стадии миграции: маргинация, адгезия, эмиграция Первыми эмигрируют нейтрофилы → моноциты (через 24-28 ч) → лимфоциты и др. (через 72 ч и более)
Медиаторы воспаления → экспрессия на эндотелиоцитах молекул адгезии (селектины, интегрины, молекулы, относящиеся к семейству Ig) → взаимодействие эндотелиоцитов с L → взаимодействие L с межклеточным матриксом → накопление L в очаге воспаления
Стадия адгезии опосредуется селектинами: среди селектинов выделяют L-селектин (экспресс. постоянно на пов-ти всех L), E-селектин (экспресс. на мембранах эндотелиоцитов после их активации цитокинами), Р-селектин (синтезируется в эндотелиаоцитах постоянно и накапливается в секреторных тельцах)
Медиаторы воспаления (гистамин, ИЛ-1, ФНО, ЛПС м/о) → появление Р-селектина на поверхности клеток → взаимодействие с лигандами на пов. L → первоначальное замедление и rolling L – «качение» по эндотелию (позже в процесс вмешивается L-селектин) → ↑ t контакта L с эндотелием → ↑ вероятности активации L медиаторами воспаления → появление на поверхности L интегринов, которые обеспечивают более плотное прикрепление L к эндотелию (лиганды интегринов – ICAM-1, ICAM-2, VCAM-1 на поверхности эндотелиоцитов) → миграция L во внесосудистом пространстве (L «цепляются» с помощью интегринов за элементы межклеточного матрикса: фибронектин, ламинин, коллаген) → проникновение в очаг воспаления → фагоцитоз
Особую роль в процессе эмиграции L играют хемоаттрактанты (ХА):
·активируют L
·мобилизуют внутриклеточные органеллы L и вызывают хемотаксис (активное перемещение
внаправлении наибольшей [ХА])
Наиболее сильные ХА, воздействующие на разные формы L:
·ЛПС м/о (входят в состав клеточной оболочки Грбактерий)
·фрагменты комплемента С5а-С9
·ЛТ В4 (а вообще большинство ЛТ)
·фактор активации тромбоцитов
Хемокины – ХА, воздействующие преимущественно на специальные подтипы L (низкомолекулярные белки, которые секретируются различными участвующими в воспалительном ответе клетками)
·ИЛ-8 (ХА для нейтрофилов)
·MCP-1 (ХА для моноцитов)
·лимфотактин (ХА для лимфоцитов и NK)
·эотаксин (ХА для эозинофилов)
ХА → специфические R на пов-ти L → ↑ [Ca2+] в цитоплазме → активация микротубулярной системы (образующей внутренний скелет) → появление псевдоподий + активация ФЛ А2 (ПКа, ПКс) → арахидоновая кис-та → образование Pg и ЛТ (участвуют в воспалении)
Фагоцитоз – процесс узнавания, активного захвата и поглощения микроорганизмов, разрушенных клеток и инородных частиц специализированными клетками иммунной системы Облигатные фагоциты: макрофаги (моноциты, МФ), микрофаги (нейтрофилы, эозинофилы, базофилы)
Стадии фагоцитоза: прилипание, поглощение, внутриклеточное разрушение
Прилипание – обусловлено существованием на мембране фагоцитов R для молекул (входящий в состав микробной клетки/появляющихся на поверхности собственной погибающей клетки), но в большинстве случаев прилипание фагоцитов осуществляется при участии опсонинов Опсонины – сывороточные фак-ры, попадающие в очаг воспаления в составе воспалительного экссудата (Ig, фрагмент C3b, некоторые плазменные белки)
Молекулы опсонинов располагают двумя участками, один из которых связывается с поверхностью атакуемой частички, а другой — с мембраной фагоцита (например, IgG связывается Fab-фрагментом с АГ м/о, а Fc-фрагментом – с мембраной фагоцитов, на которой есть R для Fc-фрагмента) Поглощение: образование псевдоподий фагоцитом → окружение объекта → объект оказывается
«внутри мешка» - фагосомы → внутриклеточное разрушение
При незавершенном фагоцитозе, гибели L, разрушении мембраны фагосомы → выход содержимого гранул и активных метаболитов кислорода → гибель объекта атаки + повреждение тканей организма
Доп. инфа про бактерицидные системы: https://docs.google.com/document/d/1Sm9h24vqZPWpgY7dOmXMVqb3TK9l1q4DCuDOgZHL23Q/edit
24. Диалектика защиты и повреждения в процессе развития воспаления.
Как и всякий патологический процесс, воспаление по своей сущности процесс противоречивый. В нем сочетаются и мобилизация защитных сил организма, и явления повреждения («полом»). Возникнув в филогенезе как явление приспособительное, воспаление сохранило это свойство и у высших животных. Организм защищается от воздействия чуждых и вредных ему факторов путем отграничения воспалительного очага от всего организма. Такое действие предотвращает распространение и генерализацию воспалительного процесса, сосредоточивая борьбу с вредным агентом в одном очаге. Воспаленная зона не только фиксирует все, что происходит в ней, но и поглощает токсические вещества, циркулирующие в крови. Это объясняется формированием вокруг очага воспаления своеобразного барьера с односторонней проницаемостью. Вначале такой барьер создается путем закупорки отводящих лимфатических и кровеносных сосудов и блокады внесосудистого тканевого транспорта. Далее он окончательно формируется вследствие размножения соединительнотканных клеток на границе между здоровой и пораженной тканью. Защитная роль воспалительного барьера наглядно демонстрируется в эксперименте со стрихнином, смертельная доза которого не приводит к гибели животного, если ее ввести в воспалительный очаг.
В очаге воспаления создаются неблагоприятные условия для жизни микроорганизмов. В этом отношении главную роль играют фагоциты и специфические антитела, а также ферменты и основные белки. Целебные свойства воспаления особенно отчетливо проявляются в стадии пролиферации и регенерации. Однако все изложенное выше отражает только одну (положительную) сторону воспаления. Вторая, противоположная, заключается в том, что воспаление всегда несет в себе элемент разрушения. Борьба с «агрессором» в зоне воспаления неизбежно сочетается с гибелью собственных клеток. В некоторых случаях начинает преобладать альтерация, что приводит к гибели ткани или целого органа. Экссудация может вызвать нарушение питания ткани, ее ферментативное расплавление, гипоксию и общую интоксикацию. И. И. Мечников отмечал, что «целительная сила природы, главный элемент которой составляет воспалительная реакция, вовсе не есть еще приспособление, достигшее совершенства».
Представление о воспалении, как о патологическом процессе, в котором «защитительное» и «собственно патологическое» находятся в единстве и борьбе, соответствует действительности.
25. Локализация и генерализация повреждения. Местные и общие реакции на
повреждение. Их взаимосвязь.
Альтерация (повреждение) – касается всех структур ткани, попавших под воздействие повреждающего фактора (флогогена), вызывающего необратимые и обратимые изменения клеток, межклеточного вещества, нервных окончаний, сосудов.
Повреждающие факторы (флогогены) → повреждение → комплексная интегративная реакция, включающая в себя:
1)Местный типовой процесс – воспаление