

Патофизиология (Пособие для резидентуры)
.pdfРОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ |
4 |
превращение фенилаланина в тирозин, в крови и моче накапливаются фенилаланин и продукты его катаболизма – фенилпировиноградная, фенилмолочная и фенилуксусная кислота. У больных возникают слабоумие и судорожные припадки. Из-за снижения содержания тирозина уменьшается синтез меланина, в результате этого уменьшается пигментация волос. При добавлении к моче больного раствора соли железа 3-хлор уксусной кислоты моча окрашивается в зеленый цвет с оливковым оттенком), муковисцидоз (протоки экзокринных желез закупориваются густым секретом. Это связано с увеличением резорбции натрия и замедлением выведения хлора из эпителиальных клеток железистых протоков. Выделяют легочную и кишечную формы. Наблюдаются бронхит, пневмония, эмфизема легких, синдром мальабсорбции, кишечная непроходимость), болезнь Вильсона-Коновалова (в результате генетического дефекта в печени нарушается соединение меди с церулоплазмином, в организме увеличивается содержание меди, которая накапливается в печени, мозге, роговице глаза) и др. (табл. 4.3).
Таблица4.3. Аутосомно-рецессивныезаболевания
Локализацияповреждения |
Заболевания |
|
|
|
|
Нервная система |
Атаксия Фридрейха |
|
Микроцефалия |
||
|
||
|
Фенилкетонурия |
|
|
Фруктозурия |
|
|
Гистидинурия |
|
|
Галактоземия |
|
Обмен веществ |
Алькаптонурия |
|
Гомоцистинурия |
||
|
||
|
Болезнь Вильсона-Коновалова |
|
|
Липидозы |
|
|
Гликогенозы (I-X тип) |
|
|
Болезнь Танджера |
|
|
Синдром Луи-Барра |
|
Иммунная система |
Синдром Глансман-Риникера |
|
|
Синдром Незелофа |
|
Слуховой и зрительный аппараты |
Врожденная глухонемота |
|
Пигментный ретинит |
||
|
||
|
Болезнь Бернард-Сулье |
|
Система крови |
Тромбастения Глансмана |
|
Гемофилия C |
||
|
||
|
Афибриногенемия |
|
Эндокринная система |
Болезнь Райли-Даи |
|
Адреногенитальный синдром |
||
|
||
Печень |
Синдром Дабин-Джонсона |
|
Синдром Ротора |
||
|
||
|
Синдром Вернера |
|
Система репарации ДНК |
Синдром Хатчинсон-Гильфорда |
|
Синдром де Санктис Каккионе |
||
|
||
|
Пигментная ксеродермия |
Особенности аутосомно-рецессивного типа наследования:
женщины и мужчины болеют с одинаковой частотой;
часто родители больного ребенка являются родственниками;
обычно родители больного фенотипически здоровы, являются гетерозиготными носителями. Вероятность рождения больных детей составляет 25%;
51

|
|
|
|
ЧАСТЬ I |
ОБЩАЯ ПАТОФИЗИОЛОГИЯ |
|
|
|
если один из родителей гетерозиготный (фенотипически здоров), другой гомозиготный (фенотипически болен), 50% детей рождаются больными, а 50% – здоровыми. Однако здоровые дети являются рецессивными носителями этого заболевания;
если один из родителей болен, дети рождаются фенотипически здоровыми.
 Наследование, сцепленное с Х-хромосомой. При этом патологический ген локали-
Наследование, сцепленное с Х-хромосомой. При этом патологический ген локали-
зуется в X хромосоме. Заболевания, наследуемые по Х-сцепленному рецессивному типу, встречаются преимущественно у мужчин, т.к. у них одна Х-хромосома. Это является причиной фенотипических изменений при мутации этой хромосомы. Т.к. у женщин две Х-хромосомы, эти наследственные заболевания проявляются только в гомозиготном состоянии. Это встречается в редких случаях, т.к. эти мутации в гомозиготном состоянии в большинстве случаев летальные. Чаще встречается X-сцепленное рецессивное наследование. Поэтому женщины будучи фенотипически здоровыми, являются носительницами патологического признака.
К заболеваниям, наследуемым по X-сцепленному рецессивному типу, относятся: гемофилия А (антигемофильный глобулин не синтезируется) и гемофилия В, дальтонизм (цветовая слепота), синдром Леш-Нихана (нарушение пуринового обмена, сопровождающееся гиперурикемией), юношеская глаукома, болезнь Менкеса (возникает в результате мутации гена, кодирующего синтез белка, транспортирующего медь в печень. Это приводит к уменьшению содержания меди в сыворотке крови, печени, головном мозге. Волосы становятся депигментированными, кучерявыми, возникают тяжелые повреждения ЦНС), гликогеноз XI типа (возникает в результате недостаточности фосфогексоизомеразы, сопровождается накоплением гликогена в печени и эритроцитах) (табл. 4.4).
Таблица4.4. Х-сцепленныерецессивныеболезни
Локализацияповреждения |
Заболевания |
|
|
|
|
Обмен веществ |
Несахарный диабет |
|
Синдром Леш-Нихана |
||
|
||
|
|
|
|
Гемофилия А и B |
|
Система крови |
Хроническое гранулематозное заболевание |
|
|
Недостаточность глюкоза-6-фосфатдегидрогеназы |
|
|
|
|
Опорно-двигательная система |
Мышечная дистрофия Дюшенна-Беккера |
|
|
|
|
|
Агаммаглобулинемия (синдром Брутона) |
|
Иммунная система |
Селективная недостаточность IgA |
|
|
Синдром Вискотт-Олдрича |
|
|
|
Особенности передачи заболеваний, наследуемых по X-сцепленному рецессивному типу:
патологический ген передается от отца дочерям (сыновьям не передается), дочери больного отца фенотипически здоровые носительницы патологического гена;
вероятность рождения больного ребенка от женщины-носительницы 25% (50% родившихся мальчиков больные);
больной мужского пола получает патологический ген только от матери;
носительницыпатологического гена могутунаследоватьпатологический ген и ототца, и от матери;
52

РОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ |
4 |
женщины болеют в редких случаях. Только дочь от брака гемизиготного отца и гетерозиготной матери может быть больна. При этом заболевание протекает тяжело. Новорожденныйребенок, какправило, недоживаетдо одногогода.
Некоторые моногенные заболевания наследуются согласно всем трем типам наследо-
вания закона Менделя. В качестве примера можно привести синдром Элерса-Данлоса. Заболевание развивается в результате генетического дефекта синтеза коллагена. Поэтому это заболевание относится с синдромом Марфана к одной и той же категории. Синдром ЭлерсаДанлоса передается из поколения в поколение по аутосомно-доминантному, аутосомнорецессивному и по сцепленному с Х хромосомой типам наследования. Это связано с разнообразием белков, участвующих в синтезе коллагена. К основным признакам относятся склонность к кровотечениям, деформации в скелете, гиперэластичность кожи.
Сцепленный с X-хромосомой доминантный тип наследования. Витамин D-резис-
тентный рахит, темно-коричневая зубная эмаль, фолликулярный гиперкератоз (полное или частичное выпадение ресниц, бровей) и др. – примеры патологий с этим типом наследования. Особенности передачи заболеваний, наследуемых по доминантному типу сцепленному с X хромосомой:
болеют лица и мужского, и женского пола. Однако больных женщин в 2 раза больше, чем больных мужчин;
больная женщина передает патологический ген половине своих дочерей и сыновей;больной мужчина передает патологический ген только всем дочерям, но не сыновьям.
Сыновья получают от отца только Y хромосому.
Сцепленный с Y хромосомой тип наследования. Заболевания с этим типом наследо-
вания передаются от отца всем сыновьям. К таким заболеваниям относится гипертрихоз (избыточный рост волос на теле).
4.4. Наследственныеболезни, неподчиняющиесязаконуМенделя
Известно, что некоторые наследственные болезни передаются не по закону Менделя. Основу наследования таких заболеваний составляют импринтинг, повторение триплетов в геноме. Помимо этого существуют наследственные болезни, передающиеся митохондриальными генами.
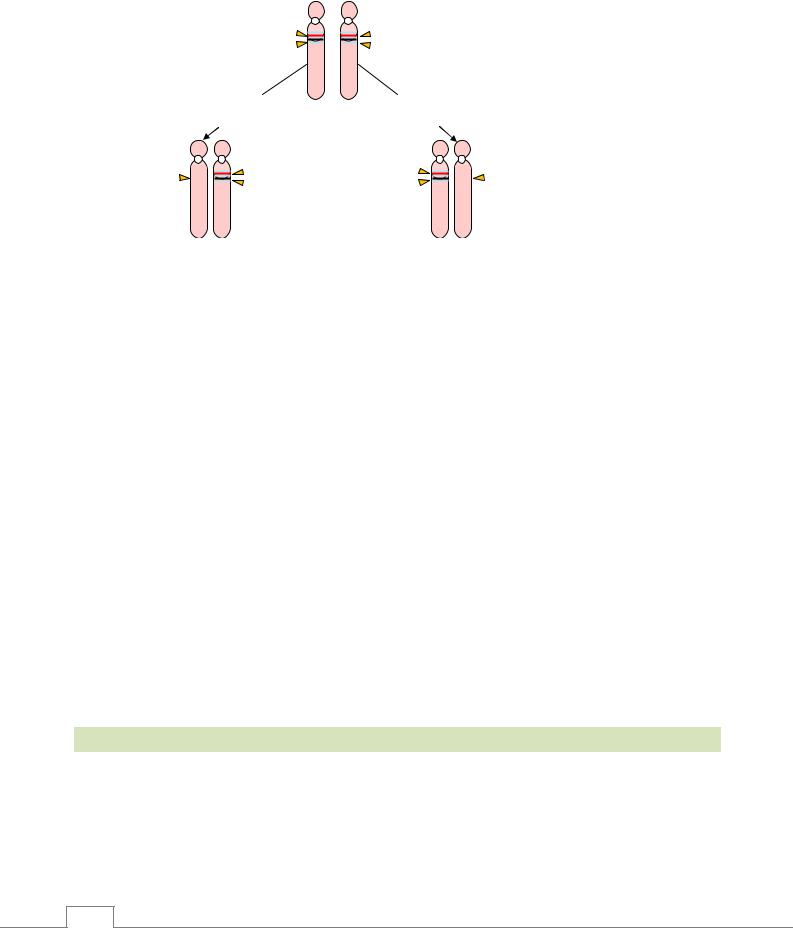
Заболевания, передающиеся по импринтингу. В норме один из гомологичных генов,
наследуемых от родителей, может быть активный, а другой неактивный (импринтинг). Например, ген Энгельмана, расположенный в определенном локусе 15 хромосомы матери, активный, а ген Прадера-Вилли неактивный; у отца, наоборот, в том же локусе расположенный ген Прадера-Вилли активный, а ген Энгельмана неактивный (рис. 4.1). В активных генах по разным причинам может произойти мутация. В зависимости от того, от кого из родителей передается мутировавший активный ген, возникают различные наследственные заболевания. Это называется феномен импринтинга. К заболеваниям, связанным с феноменом импринтинга, относятся синдромы Прадера-Вилли и Энгельмана.
Синдром Прадера-Вилли возникает в результате делеции гена Прадера-Вилли (активный ген) 15-ой хромосомы, унаследованной от отца. При этой патологии наблюдаются умственная отсталость, низкорослость, мышечная гипотония, выраженное ожирение, гипогонадизм и другие признаки.
53
|
|
|
|
ЧАСТЬ I |
ОБЩАЯ ПАТОФИЗИОЛОГИЯ |
|
|
|
|
Материнская хромосома |
Отцовская хромосома |
|
|
Неактивный ген Прадера-Вилли |
Активный ген Прадера-Вилли |
|||
Активный ген Энгельмана |
||||
Неактивный ген Энгельмана |
||||
|
Делеция участка на |
Делеция участка на |
|
|
|
материнской хромосоме |
отцовской хромосоме |
|
|
|
Активный ген |
Неактивный ген |
|
|
Участок |
Прадера-Вилли |
Прадера-Вилли |
Участок |
|
делеции |
|
|
||
Неактивный ген |
Активный ген |
делеции |
||
|
||||
|
Энгельмана |
Энгельмана |
|
|
СИНДРОМ ЭНГЕЛЬМАНА |
СИНДРОМ ПРАДЕРА-ВИЛЛИ |
|||
Рис. 4.1. Синдромы Энгельмана и Прадера-Вилли /9/.
Синдром Энгельмана (“счастливая кукла”) возникает в результате делеции гена Энгельмана (активный ген) 15-ой хромосомы, унаследованной от матери. У таких больных наблюдаютсяслабоумие, неадекватноесчастливое выражениелицасулыбкой.
Митохондриальные заболевания – передаются по наследству только по материнской линии. Патология встречается у всех детей больной матери. Все дети больного отца и здоровой матери бывают здоровыми. Т.к. в зиготе от 0 до 4 митохондрий, наследуемых от отца, и до 2500 митохондрий, наследуемых от матери. После оплодотворения репликация отцовских митохондриальных ДНК блокируется. По этому типу наследуются синдромы Лебера (атрофия зрительного нерва) и Лея (митохондриальная энцефалопатия), миоклональная эпилепсия, семейнаядилатационная кардиомиопатияидр.
Экспансия повторения триплетов (экспансия тринуклеотидных повторов). В
норме в некоторых генах число повторения триплетов может колебаться в определенных пределах. Если число повторения триплетов превышает определенный предел, это приводит к нарушению функции этого гена и становится причиной развития заболевания. К таким мутациям относятся синдром «фрагильной (ломкой) Х хромосомы» или синдром МартинаБелла /7/. Для этой группы заболеваний характерны следующие признаки:
признаки экспансии повторения триплетов проявляются поздно, сопровождается нейродегенеративными заболеваниями;
прямая связь между числом повторения триплетов и клинической картиной. Для этих заболеваний характерна генетическая антиципация, т.е. в последующих поколениях заболевание встречается в более тяжелой форме.
4.5. Хромосомныеболезни
Заболевания, сопровождающиеся изменениями количества и структуры хромосом, называют хромосомными болезнями. В результате оплодотворения гамет с дефицитом или избытком хромосом рождаются дети с хромосомными заболеваниями (синдромы Шерешевского-Тернера, Клайнфельтера, трисомия Х-хромосомы, ХYY синдром и др.). Причинный фактор большинства хромосомных болезней – геномная мутация (синдромы Дауна,
54

РОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ |
4 |
Патау, «кошачьего крика», Эдвардса и др.). Хромосомные болезни характеризуются следующими признаками:
общие признаки – слабоумие, психические расстройства, бесплодие, короткая продолжительность жизни (больные с аномалиями половых хромосом имеют относительно большую продолжительность жизни);
аномалии головы, лица, верхних и нижних конечностей – микроцефалия, сужение глазных щелей, неполное окостенение, деформации скелета и т.д.;
пороки развития внутренних органов – пороки развития сердца и мочеполовой системы
ит.д.
Хромосомные болезни подразделяются на две группы: половые и соматические. К аномалиям половых хромосом относятся:
Синдром Шерешевского-Тернера
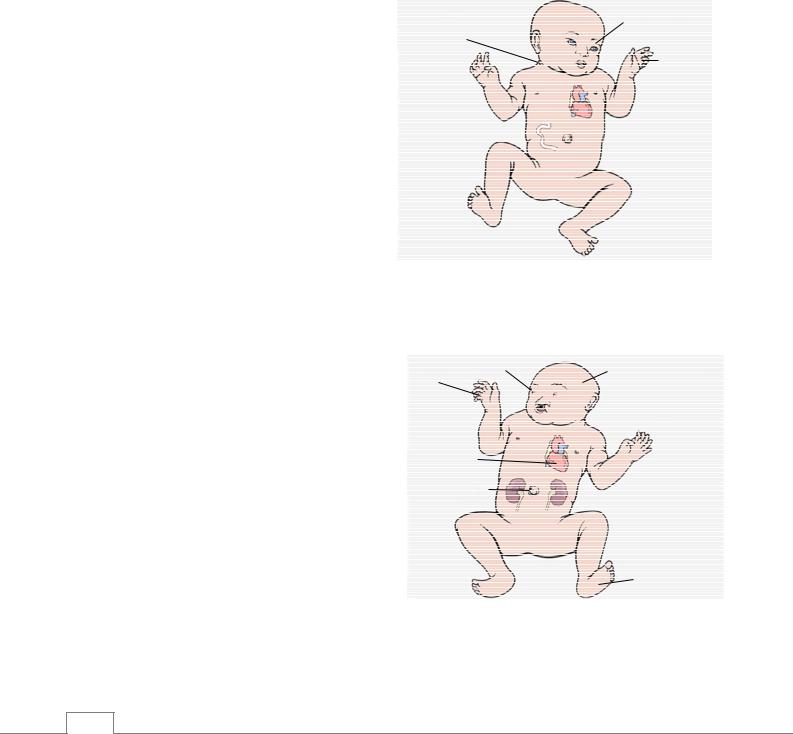
(моносомия Х-хромосомы) – в кариотипе таких больных одна Х-хромосома (45,X) (рис. 4.2), а число аутосомных хромосом – в норме. Наличие Х-хромосомы в кариотипе обуславливает развитие женского пола. У больных низкий рост (120-140 см), отставание от развития половых органов (инфантильное состояние), гипоплазия или полное отсутствие развития яичников, бесплодие. Часто у таких больных широкая крыловидная складка на шее (pteriqium coli). Наблюдаются лимфатические отеки рук и ног, врожденные пороки сердца, сужение аорты, высокое расположение небной линии. При синдроме Шерешевского-Тернера в эпителиальных клетках слизистой оболочки внутренней поверхности щек половой хроматинне обнаруживается.
Низкое расположение линии роста волос на задней стороне шеи
Крыловидная
складка
Широкая грудная клетка и широко расставленные грудные соски
Пигментные пятна
Низкорослость
Коарктация аорты
Искривленные локти
Аномалии яичников бесплодие, аменорея
Врожденный
периферический лимфатический отек
Рис. 4.2. Синдром Шерешевского-Тернера /9/.
Синдром Клайнфельтера – при этом заболевании в кариотипе 47 хромосом (47, XXY). Наличие в кариотипе Y хромосомы обуславливает развитие мужского пола. У таких больных высокий рост, азооспермия, бесплодие, незначительное слабоумие, астеническое телосложение (конечности относительно туловища непропорционально длинные), слабое развитие мышц. Больные бывают ленивыми и вялыми. Мальчики с синдромом Клайнфельтера до периода полового созревания нормально растут и развиваются. В период полового созревания происходит феминизация: появляются признаки, характерные для женского скелета, гинекомастия (увеличение размеров молочных желез). Определение полового хроматина в эпителиальных клетках слизистой оболочки внутренней поверхности щек позволяет подтвердить диагноз (у здоровых мужчин половой хроматин не выявляется). Встречаются также XXXY, XXXXY варианты кариотипаэтогосиндрома.
Трисомия X хромосомы – общее число хромосом 47, (кариотип 47, XXX), 3 половые хромосомы, 2 тельца Барра. У женщин с этим синдромом наблюдаются незначительное слабоумие, недоразвитие яичников, гипоплазия матки, бесплодие.
55
|
|
|
|
ЧАСТЬ I |
ОБЩАЯ ПАТОФИЗИОЛОГИЯ |
|
|
|
К заболеваниям, связанным с аномалиями аутосомных хромосом, относятся синдромы Дауна, Эдвардса, Патау и «кошачьего крика».
Синдром Дауна – встречается у детей обоего пола (кариотип: 47,ХХ,+21). Выделяют 2 цитогенетических варианта – трисомия (геномная мутация) и транслокация (хромосомная мутация). 95% больных имеют трисомию по 21 хромосоме (в результате нерасхождения гомологичных хромосом при мейозе). При варианте трисомии в кариотипе бывает 47, а при варианте транслокации 46 хромосом. При транслокации участок 21-ой хромосомы отделяется и присоединяется к другой хромосоме (14-ой или 22-ой хромосоме). Чаще наблюдается у позднородящих женщин (после 37-40 лет). Эту патологию также объясняют старением яйцеклетки и сперматозоида. Такие дети низкорослые, имеют короткие пальцы рук и ног, незначительно уплощенное лицо с раскосыми глазами (как у монголоидной расы, поэтому
раньше это заболевание называли монголизмом – mongolism), выраженные скулы, ротунихвсегдаполуоткрыт.
При синдроме Дауна наблюдаются различные физические пороки, слабоумие. Для таких больных характерны врожденные пороки сердца, четырехпалость или глубокая поперечная складка ладони («обезьянья складка»), наличие пятен на радужке и эпикантус (вертикальная кожная складка у внутренного угла глаза). Больные склонны к лимфолейкозу. На фоне синдрома Дауна высокий риск развития болезни Альцгеймера (pис. 4.3).
Слабоумие |
|
|
|
|
|
Эпикантус |
||
|
||||||||
Кожная |
|
|
|
|
плоское лицо |
|||
|
|
|
|
|
|
|||
складка шеи |
|
|
|
|
|
Обезьянья |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
складка |
Врожденные |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
пороки сердца |
|
|
|
|
|
|
||
Стеноз |
|
|
|
|
|
|
Пупочная |
|
кишечника |
|
|
|
|
|
грыжа |
||
|
|
|
|
|
|
|
|
Склонность |
|
|
|
|
|
|
|
|
к лейкозам |
Щель между первым и вторым пальцами
вторым пальцами
Рис. 4.3. Синдром Дауна /9/.
Синдром Патау – возникает при трисомии 13-ой хромосомы (кариотип: 47,ХХ,+13) или транслокации Робертсона (передача длинного плеча дополнительной хромосомы) (pис. 4.4). Для таких больных характерны выраженное слабоумие, судороги, полидакти-
лия, расщелина губы |
и неба, |
микроце- |
Микрофтальмия |
Микроцефалия и |
|||||||
фалия, микрофтальмия, анофтальмия, |
Полидактилия |
||||||||||
|
|
слабоумие |
|||||||||
крипторхизм, глухота, гемангиомы на лице |
|
|
|
|
Расщелина губы и |
||||||
и руках, пупочная и паховая грыжи, |
|
|
|
|
|||||||
|
|
|
|
неба |
|||||||
нарушения зрения и сердечно-сосудистые |
|
|
|
|
|
||||||
патологии. |
|
|
|
|
|
Пороки сердца |
|
|
|
||
Синдром “кошачьего крика” – наб- |
|
|
|
||||||||
людается |
при укорочении |
одной хромо- |
Пупочная грыжа |
|
Аномалия почек |
||||||
сомы в 5-ой паре хромосом. Больные низ- |
|
|
|
||||||||
|
|
|
|
|
|||||||
корослые, круглолицые, косоглазые. У них |
|
|
|
|
|
||||||
развиваются |
пороки |
различных |
органов |
|
|
|
|
|
|||
(сердца, половых органов, почек), син- |
|
|
|
|
|
||||||
дактилия |
пальцев нижних |
конечностей. |
|
|
|
|
Деформация |
||||
Ребенок с этим синдромом плачет голосом, |
|
|
|
|
ног (стоп) |
||||||
Рис. 4.4. Синдром Патау /9/. |
|||||||||||
напоминающим кошачий крик. Путем |
|||||||||||
проведения |
цитогенетического |
анализа |
|
|
|
|
|
||||
можно поставить больному правильный диагноз. |
|
|
|
||||||||
Синдром |
Эдвардса – возникает при |
трисомии 18-ой пары |
хромосом (кариотип: |
||||||||
47,ХХ,+18). 75% таких детей редко доживают до 6 месяцев. У больных детей наблюдаются
56

РОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ |
4 |
аномалии черепа, лица, короткая грудная кость, узкие межреберные пространства, короткая и широкая грудная клетка, отставание умственного развития (рис. 4.5).
Фенокопии, имеющие сходные с |
Выпуклый холм Слабоумие |
||||||||
наследственной |
патологией |
фенотипические |
|
|
|
|
|
|
|
клинические проявления, но в отличие от нее, |
Низкое расположение ушных |
|
Микрогнатия |
||||||
эти синдромы развиваются в результате |
раковин |
|
|||||||
|
|
||||||||
влияния факторов внешней среды в |
Короткая шея |
|
|
||||||
эмбриональном периоде. К наследственным |
|
|
|
|
|
|
|
||
заболеваниям, |
имеющим |
аналогичные |
Деформация пальцев |
|
|
||||
фенокопии, относятся: |
|
|
|
|
|
|
|
|
|
гаргоилизм (болезнь Гюнтера-Пфаунд- |
Врожденные |
|
|
||||||
лера) – аутосомно-рецессивное или сцеп- |
пороки сердца Аномалия |
|
|
|
|
|
|||
|
|
|
|
|
|||||
ленное с Х-хромосомой наследственное |
почек |
|
|
||||||
заболевание. |
Характеризуется накопле- |
Ограничение подвижности в |
|
|
|
|
|
||
|
|
|
|
||||||
нием в тканях ганглиозидов (циалоглико- |
тазобедренном суставе |
|
|
||||||
|
|
|
|
|
|
|
|||
липидов). У таких больных низкий рост, |
|
|
|
|
|
|
|
||
большая и уродливая голова. У них |
|
|
|
|
|
|
|
||
наблюдаются глухота, катаракта, кифоз. |
|
|
|
|
|
|
|
||
Фенокопия |
этого заболевания встреча- |
Деформация |
|
|
|
|
|||
ется у детей, родители которых страдают |
|
|
|
||||||
|
|
|
|||||||
ног (стоп) |
|
|
|||||||
хроническим алкоголизмом; |
Рис. 4.5. Синдром Эдвардса. |
||||||||
|
|
|
|||||||
 ранняя глухота – как признак передается рецессивным или доминантным путем. Однако как фенокопия может встречаться и у детей, мать которых в период беременности перенесла краснуха;
ранняя глухота – как признак передается рецессивным или доминантным путем. Однако как фенокопия может встречаться и у детей, мать которых в период беременности перенесла краснуха;  врожденная катаракта – передается по аутосомно-доминантному типу
врожденная катаракта – передается по аутосомно-доминантному типу
наследования. Однако может встречаться и фенокопия этого заболевания у детей, мать которых в период беременности перенесла краснуха или подвергалась облучению.
4.6. Методыизученияидиагностикинаследственныхболезней
К генетико-эпидемиологическим методам относятся:
Генеалогический метод – основан на изучении родословной. Это основной метод исследования генетических заболеваний.
Близнецовый метод – изучает степень влияния наследственности и среды на развитие какого-либо нормального или патологического признака у однояйцовых (монозиготных) и двуяйцовых (дизиготных) близнецов. В этих исследованиях отмечают схожесть (конкордантность) и различие (дискордантность) признаков у обоих близнецов. Конкордантность – это проявление определенного признака у обоих сравниваемых близнецов. Дискордантность – это отсутствие у одного из близнецов признака, имеющегося у другого /2/.
Демографо-статистический метод – изучает распространенность наследственных заболеваний в различных географических зонах.
Клинические методы диагностики. Основная цель – это выявление с помощью специальных диагностических методов наследственного характера патологий.
Цитогенетический (кариологический) метод – с помощью проведения микроскопи-
ческих исследований изучается число и особенности строения хромосом клетки (кариотип). Этот метод имеет большое значение в диагностике хромосомных болезней.
Молекулярно-генетический метод – изучается последовательность расположения отдельных нуклеотидов в ДНК.
Биохимический метод – используется в изучении нарушений метаболизма, носящих наследственный характер. При этом применяются биохимические маркеры, специфические
57

|
|
|
|
ЧАСТЬ I |
ОБЩАЯ ПАТОФИЗИОЛОГИЯ |
|
|
|
для наследственных заболеваний. Например, для подтверждения диагноза фенилпировиноградной олигофрении определяется количество фенилпировиноградной кислоты в моче.
Иммунологический метод – используется в диагностике агаммаглобулинемии, дисгаммаглобулинемии, атаксии-телеангиэктазии, антиген-антитело несовместимости между организмом матери и плода.
Метод определения полового хроматина – половой хроматин, будучи одной из Х-хро-
мосом, в процессе дифференциации уплотняется (инактивируется) и приобретает способность интенсивно окрашиваться. Половой хроматин был впервые открыт в 1949 году М.Барром в мозговых клетках кошки. Этот метод используется в диагностике синдромов Шерешевского-Тернера, Клайнфельтера, трисомии Х-хромосомы.
Метод дерматоглифики – основывается на изучении ладонной поверхности руки. Например, при синдроме Дауна в 40% случаев на коже ладонной поверхности руки больного выявляется глубокая поперечная складка («обезьяная складка»). Другой признак синдрома Дауна, встречающийся в 20% случаев – наличие на внутренней поверхности мизинца лишь одной складки.
Экспериментальный метод (биологическое моделирование) – на животных создаются модели наследственных болезней. Например, путем моделирования гемофилии на собаках, ахондроплазии на кроликах, атеросклероза на голубях, мышечной дистрофии на мышах и курицах изучаются вышеуказанные наследственные заболевания.
4.7. Основныепринципылечениянаследственныхболезней
Лечение наследственных болезней проводится в трех направлениях: этиологическое, патогенетическое, симптоматическое.
Этиологическое лечение направлено на устранение причины, вызывающей заболевание. С этой точки зрения «генной инженерии» придается большое значение. Основная цель – замена мутантного«больного» гена «здоровым» геномв геномеклеткиповрежденного органа.
Патогенетическое лечение проводится с целью устранения основного звена патогенеза
иостановки развития болезни. Для этого применяют несколько методов /7/:
замещающая терапия – введение в организм недостающих веществ. Например, введение при гемофилии антигемофильного глобулина, при гликогенозах – соответствующего фермента и т.д.;
коррекция метаболизма:
a)предотвращение поступления в организм метаболически плохо усваиваемых
веществ (например, фенилаланина и лактозы); б) удаление накопившихся в организме метаболитов (например, фенилпировино-
градной кислоты или холестерина); в) регуляция активности ферментов (например, при миодистрофиях снижение
активности креатинфосфокиназы, при гиперхолестеринемии повышение активности липопротеинлипазы);
хирургическая коррекция дефектов. Например, при гликогенозе печени наложение анастомоза между нижней полой веной и воротной веной (портокавального шунта). Это приводит к тому, что часть глюкозы, усвоенной в кишечнике, не попадая в печень, поступает в общий кровоток и распространяется в организме. Таким образом, предотвращается накопление гликогена в печени.
Симптоматическое лечение направлено на устранение признаков, осложняющих состояние больного. Например, при полидактилии – удаление дополнительного пальца, при дефектах опорно-двигательной (костно-суставной) системы, в области сердца, лица – устранение дефектов хирургическим путем, лечение ряда наследственных заболеваний нервной системы физическими методами (климатотерапия, тепловые процедуры и др.).
58

РОЛЬ КОНСТИТУЦИИ ОРГАНИЗМА В ПАТОЛОГИИ |
5 |
Глава5. РОЛЬКОНСТИТУЦИИОРГАНИЗМАВ ПАТОЛОГИИ
5.1. Общеепонятиеоконституцииорганизма, классификация конституциональныхтипов
Конституция (“constitutio” – строение) – это комплекс наследственных и приобретенных, устойчивых морфологических и функциональных свойств организма. Конституциональные особенности организма влияют на индивидуальную реактивность, способность к адаптации, течение физиологических и патологических процессов.
Конституциональные типы классифицируют по разным принципам. Первая классификация по темпераменту человека была дана Гиппократом /5/:
сангвиники – эмоциональные, общительные, дружелюбные, активные, энергичные, взрывные, быстро принимающие правильные решения. Они способны длительно выполнять напряженную работу;
флегматики – спокойные, с высокой трудоспособностью, устойчивы к непатогенным и патогенным воздействиям. Однако на раздражители флегматики отвечают медленнее, чем сангвиники;
холерики – отличаются неуравновешенностью и нервозностью. Несмотря на то, что у них высокая трудоспособность, они быстро устают;
меланхолики – необщительные, замкнутые и нерешительные, с низкой трудоспособностью.
Классификация конституциональных типов по морфологическим признакам принадлежит французскому ученому Д.Р.Сиго. По Сиго выделяют 4 конституциональных типа:
дыхательный, или респираторный тип – характеризуется длинной грудной клеткой;
пищеварительный, или дигестивный тип – характеризуется большими размерами живота и хорошо развитой нижней 1/3 частью лица (развитый жевательный аппарат), короткой шеей;
мышечный, или мускульный тип – характеризуется развитой скелетной мускулатурой, широкой грудной клеткой, квадратной формой лица;
мозговой, или церебральный тип – отличается от других типов большим черепом, стройным телосложением, слабым развитием мышц.
Немецкий психиатр Э.Кречмер показал наличие взаимосвязи между внешними
морфологическими особенностями, психикой и психическими болезнями организма. Он выделял 3 конституциональных типа: атлетический, пикнический и астенический.
Атлетический тип соответствует мышечному, пикнический – пищеварительному, а астенический – дыхательному типу классификации по Сиго. По мнению Э.Кречмера у людей атлетического типа чаще встречается эпилепсия, астенического типа – шизофрения, пикнического типа – маниакально-депрессивный психоз.
В медицинской практике широко распространена классификация М.В.Черноруцкого, основанная на особенностях телосложения, функций и метаболических процессов. По Черноруцкому выделяют 3 конституциональных типа:
гипостенический, или астенический тип – людей этого типа отличают высокий рост,
удлиненная форма грудной клетки, длинные конечности, относительно небольшой
59

|
|
|
|
ЧАСТЬ I |
ОБЩАЯ ПАТОФИЗИОЛОГИЯ |
|
|
|
размер сердца и паренхиматозных органов, тонкая и бледная кожа, слабо развитый подкожный жировой слой, низкое расположение диафрагмы. У астеников низкое артериальное давление, большая жизненная емкость легких, слабые двигательная и секреторная функции желудка, низкая всасывающая функция кишечника, ускоренный обмен веществ, в крови низкое содержание эритроцитов, гемоглобина, холестерина, кальция, мочевой кислоты и глюкозы. При этом наблюдается гиперфункция щитовидной железы и гипофиза и гипофункция надпочечников и половых желез. У них чаще наблюдаются язва желудка и 12-перстной кишки, хронический колит;
гиперстенический тип – средний рост, относительно короткие грудная клетка и конечности, за исключением легких большие размеры внутренних органов, высокое расположение диафрагмы отличают этот конституциональный тип. У них наблюдается высокое артериальное давление, низкий обмен веществ, низкая толерантность куглеводам, активные двигательная и секреторная функции желудка, высокая всасывающая функция кишечника, в крови высокое содержание эритроцитов, гемоглобина, холестерина. При этом наблюдается гиперфункция надпочечников, половых желез и гипофункция щитовидной железы. У гиперстеников чаще встречаются сердечно-сосудистые патологии (атеросклероз, гипертония, инфаркт миокарда), желчнокаменная болезнь, сахарный диабет;
нормостенический тип – занимает промежуточное положение между гипо- и гиперстеническим типами и отличается пропорциональностью размеров тела и его частей. У нормостеников наблюдается предрасположенность к заболеваниям верхних дыхательных путей и опорно-двигательного аппарата.
А.А.Богомолец в зависимости от строения и функций соединительной ткани выделяет следующие конституциональные типы:
астенический тип – эти лица имеют слабо выраженную соединительную ткань;
фиброзный тип – имеют жесткую и волокнистую соединительную ткань;
липоматозный тип – преимущественно развита жировая ткань;
пастозный тип – у них преимущественно рыхлая соединительная ткань.
И.П.Павлов в основу классификации конституциональных типов положил деятельность высшей нервной системы, т.е. особенности возбуждения и торможения организма. Он выделял следующие 4 конституциональных типа:
сильный, уравновешенный, подвижный тип (соответствует сангвиникам) – они устойчивы к действию факторов внешней среды и легко приспосабливаются к изменившимся условиям внешней среды;
сильный, уравновешенный, неподвижный (инертный) тип (соответствует флегма-
тикам) – у них процессы возбуждения и торможения уравновешены, однако скорость течения этих процессов снижена;
сильный, неуравновешенный, подвижный тип (соответствует холерикам) – процессы возбуждения ярко выражены, протекают быстро, но кратковременны;
слабый тип (соответствует меланхоликам) – процессы возбуждения и торможения слабые, тяжело приспосабливаются к изменениям условий внешней среды.
По классификации Павлова, основанной на степени выраженности и превалирования первой и второй сигнальной системы высшей нервной деятельности, выделяют 4 группы людей:
первый тип – первая и вторая сигнальная система одинаково слабо проявляются;
второй тип (художественный) – первая сигнальная система составляет преимущество;
третий тип (прогрессивный тип) – вторая сигнальная система составляет преимущество;
60