

Витаминотерапия как этиотропная помощь при наследственных неврологических заболеваниях взрослого возраста
Неврология Генетика перевод витамины
Оригинал Перевод: Артур Арсеньев
Редакция: Мария Чжу Оформление: Никита Родионов Публикация: 16.06.2021

Неврологические генетические заболевания, манифестирующие в подростковом и взрослом возрасте, принадлежат к гетерогенным состояниям, обусловленным нарушением множества генов и проявляющимися крайне разнообразными фенотипами. Зачастую такие заболевания не имеют специфического лечения, однако некоторые из этих заболеваний поддаются терапии витаминами [1]. Витамины — необходимые микронутриенты, синтезируемые в организме в недостаточных количествах либо вовсе не синтезируемые, тем не менее, играющие важнейшую роль во внутриклеточной машинерии, трансформируясь в активные формы — кофакторы ферментов.
Витаминные добавки легкодоступны, обычно не вызывают выраженных побочных эффектов и эффективны, особенно на ранних этапах заболеваний. Таким образом, своевременное распознавание витамин-связанных заболеваний критически важно для неврологов.
Эта статья направлена на описание фенотипов витамин-зависимых нейрогенетических заболеваний с манифестацией в подростковом и взрослом возрасте, явно отличающихся от раннего дебюта.
Методы
Авторами статьи было определено 24 нейрогенетических заболевания, неврологические проявления которых могут возникать в возрасте от 10 лет, и характеризуются видимой реакцией на прием витаминных добавок, по крайней мере, для части пациентов.
Описываются следующие витамины: A, B1, B2, B3, B6, B8, B9, B12, E и тетрагидробиоптерин (BH4) коэнзим. Авторы выбрали именно этот возрастной порог (упоминаемый в тексте как «взрослый возраст»), поскольку, по опыту авторов, наследственные метаболические заболевания, начинающиеся в возрасте старше 10 лет, имеют фенотипы, которые похожи на фенотипы пациентов с более поздним дебютом этих заболеваний и отличаются от форм данной патологии с более ранним началом [2– 4]. Несмотря на то, что BH4 не является полноценным витамином, он представляет собой органическое соединение, действующее аналогично витаминным кофакторам [5], что объясняет решение авторов добавить BH4-ассоциированные состояния к заболеваниям, чувствительным к витаминам [6].
Обзор
Нейрогенетические заболевания, отвечающие на прием витаминов были поделены авторами статьи на 4 группы:
1.нарушение синтеза кофакторов, образующихся из витаминов;
2.нарушение транспорта (абсорбция в ЖКТ, транспорт в крови или захват клетками);
3.дефект самого энзима, кофактор которого синтезируется из витаминов;
4.вторичный витаминный дефицит (аномалии, не ассоциированные с генетическими расстройствами, вовлеченными в метаболизм витаминов).
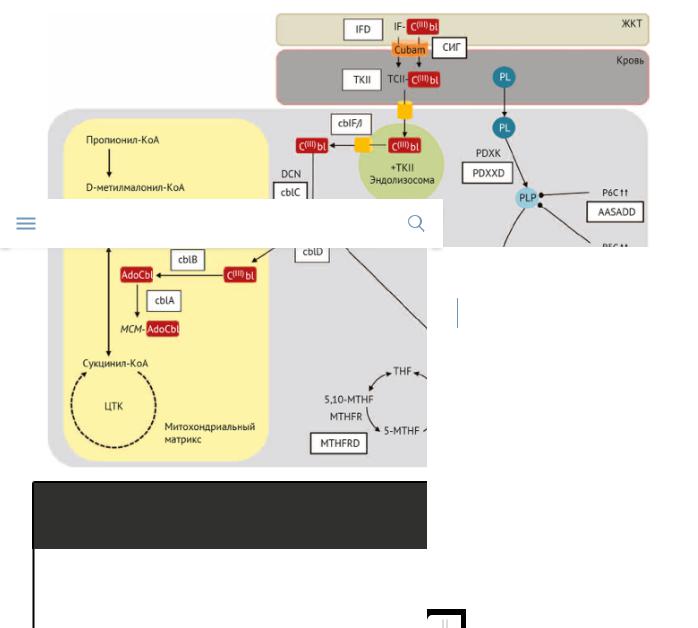
Рисунок 1 демонстрирует групповую принадлежность на примере метаболизма кобаламина.
MEDACH
Методы
Обзор
Расстройства метаболизма тиамина и поражение базальных ганглиев
Рибофлавин в мышцах и заболевания нервной системы
Разнообразные и
Рисунок 1 | Пиридоксин-, фолат- и кобаламин-ассоциированные
комплексные клинические
метаболические пути проявления дефектов обмена пиридоксина,
Все формы B12 выделены красным цветом, а B6 — голубымфолата. З болеванияи кобаламина, обусловленные нарушением ассоциированных реакций, обозначены в белых
Болезнь Хартнупа,
прямоугольниках. Патофизиология кобаламин-связанных заболеваний включает
генетический симулякр

нарушение внутриклеточного синтеза кофактором аденозилкобаламина (AdoCbl)
пеллагры
и метилкобаламина (MeCbl) из кобаламина (например, метилмалоновая ацидурия, тип cblA [cblA], метилмалоновая ацидурия и гомоцистинурияДефицит, тип cblCбиотинидазы[cblC], : метилмалоновая ацидурия и гомоцистинурия, тип cblD [cblD],имитируягомоцистинурия-
нейрооптикомиелит
мегалобластическая анемия, тип cblE [cblE]), нарушения транспорта (из просвета
тонкого кишечника, как при синдроме Имерслунд — Гресбека [СИГ], из крови к
Витамин Е и эквилибриум
тканям, как в случае синдрома дефицита транскобаламина II типа (ТКII); или внутриклеточного транспорта, например, при метилмалоновойBH4 иацидурпаркинсонизми и
гомоцистинурии, тип cblF/J), или дефекте ферментов, зависимых от витамин-
Ограничения
связанных кофакторов (при гомоцистинурии-мегалобластической анемии, тип
cblG [cblG]). Метаболизм гомоцистеина нарушен при несколькихЗаключениевитаминсвязанных нейрогенетических заболеваниях, вовлекающих витамины B6, B9, и
B12, манифестирующих во взрослом возрасте. Например, cblC обусловлен дефектом децианирования кобаламина (DCN), необходимого для катализа обратимой реакции децианирования кобаламина в полностью окисленном состоянии Co3+(C(III)bl) с переходом из лизосом в состояние Co2+(C(II)bl), с последующей конвертацией в метаболически активные формы AdoCbl и MeCbl. Аденозилкобаламин является кофактором метилмалонил-КоА-мутазы (MCM), митохондриального энзима, ответственного за конверсию метилмалонил-КоА в сукцинил-КоА, который входит в цикл трикарбоновых кислот (ЦТК). MeCbl используется как донор метильной группы для метионин-синтазы с целью генерации метионина (Met) из гомоцистеина (Hcy). Таким образом, в cblC, уровни метилмалонил-КоА и Hcy увеличиваются, а Met — уменьшаются. CblG проявляется нарушением функции энзима метионин-синтазы (MS), который используется MeCbl как кофактор. Синдром Имерслунд — Гресбека обусловлен дефицитом Cubam, тонкокишечным фактором рецептора кобаламина (IF-Cbl), состоящим из двух главных компонентов: кубилин (распознающий IF-Cbl) и амнионлесс (вовлечен в эндоцитоз IF-Cbl), кодирующимися генами кубилином (CUBN) и геном амнион-ассоциированного трасмембранного белка (AMN) соответственно. Дефицит 5,10-метилентетрагидрофолат редуктазы (MTHFRD) образуется в результате недостаточности метилентетрагидрофолат редуктазы (MTHFR), катализирующей образование 5-метилтетрагидрофолата (5-MTHF), одну из активных форм фолата; 5-MTHF является метильным донором кобаламина при снижении уровня Co+ для образования MeCbl — кофактора метионин синтазы; как в случае CblC и cblG, MTHFRD ведет к дефекту реметилирования Hcy, повышая уровень Hcy и снижая Met. Дефицит цистатионин-β-синтазы (CyBSD) включает дефицит пиридоксин-зависимого фермента цистатионин-β- синтазы (CyBS), который катализирует реакцию транссульфурации Hcy до цистатионина и, в конечном итоге, цистеина. Дефект приводит к аккумуляции Hcy и повышает его реметиляцию в Met. α-аминолипоевая-δ-семиальдегид гидрогеназы дефицит (AASADD) — является дефектом синтеза α-аминолипоевой кислоты из α-аминолипоевого-δ-семиальдегида (AASA) при метаболизме L- лизина; как результат, AASA конвертируется в 1-пиперидин-6-карбоксилат

(P6C), который ингибирует пиридоксаль 5-фосфат (PLP). Гиперпролинемия II (Hpro-II) формируется ввиду дефекта метаболизма пролина из-за дефицита 1- пирролин 5-карбоксилат (P5С) дегидрогеназы, что приводит к накоплению P5C и ингибированию PLP.
Двойные стрелки указывают на нарушение активности PLP при накоплении P5C
иP6C.
Взависимости от патофизиологии, для оптимальной эффективности применения витаминов они (или синтезируемые из них кофакторы) могут назначаться в различных фармакологических дозах и с различными путями введения. В случае протекания заболевания по вышеуказанному третьему типу, эффективность зависит от этапа вовлечения последовательных витамин-связанных реакций, часто выявляемых среди форм болезни, манифестирующих во взрослом возрасте [7–10], тогда как в случае 4 типа эффективность скорее будет слабо выражена, поскольку недостаток витаминов может представлять собой лишь эпифеномен заболевания.
Основываясь на том, что большинство фенотипов проявляются в результате различных генетических заболеваний, для более прагматичного взгляда к этим многочисленным и сложным состояниям выделяют клинико-биологический подход. Собранные данные по некоторым нейрогенетическим заболеваниям представлены на рисунке 2.
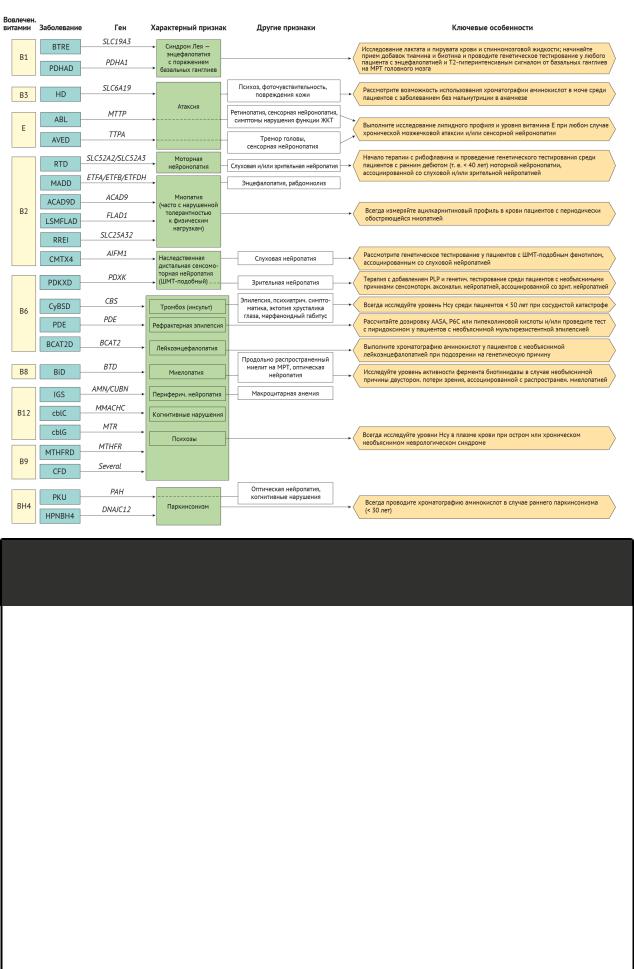
Рисунок 2 | Заболевания, отвечающие на прием витаминов, их основные клинические характеристики и ключевые аспекты
Обратите внимание, что генетическое тестирование, упомянутое в ключевых аспектах, означает исследование одного гена или клинически ориентированной генетической панели, которая должна включать данные гены.
5-MTHFR, 5-метилтетрагидрофолат; AASA, α-аминоадипин-δ-полуальдегид; ABL, абеталипопротеинемия; ACAD9, ацил-коА-дегидрогеназа 9; ACAD9D, дефицит ацил-коА дегидрогеназы 9; AIFM1, ген фактора индукции апоптоза, ассоциированный с митохондриями 1; AMN, трансмембранный белок, связанный с амнионом; ТТПА, ген белка-переносчика α-токоферола; AVED, атаксия с дефицитом витамина E; BCAT2, ген трансаминазы 2 аминокислот с разветвленной цепью; BCAT2D, дефицит трансаминазы 2 аминокислот с разветвленной цепью; BiD, дефицит биотинидазы; BTD, ген биотинидазы; BTRE, биотин-тиамин-зависимая энцефалопатия; CBS, ген цистатионин-β-синтазы; CFD, церебральный дефицит фолиевой кислоты; CMT, Шарко — Мари — Тута; CMTX4, Х-сцепленная болезнь Шарко — Мари — Тута 4 типа; CSF, спинномозговая жидкость; CUBN, ген кубилина; CyBSD, дефицит цистатионин-β-синтазы;

DNAJC12, ген C12, член семейства белков теплового шока DnaJ; ETFA/ETFB/ETFDH, гены флавопротеинов, переносящих электроны, альфа, бета и дегидрогеназы; FLAD1, ген флавинадениндинуклеотидсинтетазы 1; GI, желудочно-кишечный тракт; HD, болезнь Хартнупа; HPNBH4, гиперфенилаланинемия, легкая, без дефицита BH4; IGS, синдром Имерслунда — Грасбека; LSMFLAD, миопатия накопления липидов вследствие дефицита синтетазы FAD; MADD, множественный дефицит ацил-коА; MMACHC, метаболизм гена C, связанного с кобаламином; МRI, магнитно-резонансная томография; MTR, ген 5-метилтетрагидрофолат-гомоцистеинметилтрансферазы; MTHFR, метилентетрагидрофолатредуктаза; MTRFRD, дефицит метилентетрагидрофолатредуктазы; MTTP, ген белка-переносчика микросомальных триглицеридов; P6C, 1-пиперидин-6-карбоксилат; PDE, пиридоксинзависимые эпилепсии; PDHA1, ген субъединицы E1-α пируватдегидрогеназы; PDHAD, дефицит E1-α пируватдегидрогеназы; PDXK, пиридоксалкиназа; PDXKD, дефицит PDXK; PAH, ген фенилаланингидроксилазы; PKU, фенилкетонурия; PLP, пиридоксаль-5'-фосфат; RREI, непереносимость физических упражнений, реагирующих на рибофлавин; RTD, дефицит транспортера рибофлавина; SLC19A3, семейство 19 носителей растворенного вещества, член 3; SLC6A19, семейство 6 носителей растворенного вещества, член 19; SLC52A2/SLC52A3, семейство 52 носителей растворенного вещества, члены 2 и 3; SLC25A32, семейство 25 носителей растворенного вещества, член
32.
Расстройства метаболизма тиамина и поражение базальных ганглиев
Энцефалопатия Вернике — это приобретенное заболевание, вызванное дефицитом тиамина (витамина B1), часто в контексте недоедания и/или злоупотребления алкоголем. Энцефалопатия обычно сопровождается мозжечковыми нарушениями, судорогами, офтальмоплегией и/или птозом [11]. Схожий фенотип можно встретить при двух тиамин-ассоциированных проявляющихся во взрослом возрасте нейрогенетических заболеваниях: биотин-тиамин-зависимой энцефалопатии (BTRE) и дефиците E1-α пируватдегидрогеназы (PDHAD). Эти два состояния являются поддающимися лечению причинами генетически гетерогенного синдрома Лея (Ли) [12]. При BTRE транспорт тиамина в цитозоль снижается в результате дефекта переносчика тиамина 2-го типа (кодируется геном семейства растворенных носителей-19, член 3 [SLC19A3]), что приводит к низкому внутриклеточному уровню тиамина, тогда как уровень сывороточного тиамина остается в норме [9]. В случае PDHAD аномальная субъединица E1-α пируватдегидрогеназы (кодируемая геном субъединицы E1-α пируватдегидрогеназы [PDHA1]) препятствует правильному связыванию его кофактора тиаминпирофосфата, активного тиамина (рис. 3) [13]. Этот сбой оказывает влияние на
цикл трикарбоновых кислот с уменьшением выработки ацетил-КоА и приводит к переключению на анаэробное производство энергии и, в конечном итоге, к лактоацидозу, который также присутствует при BTRE. Помимо высокого уровня лактата, характерными чертами PDHAD являются высокие уровни пирувата в крови и спинномозговой жидкости. Как и при других метаболических заболеваниях, стрессовые состояния, такие как приступы лихорадки, хирургическое вмешательство или травмы, могут вызвать эпизоды энцефалопатии. Однако также могут возникать прогрессивно ухудшающиеся хронические симптомы, такие как дистония (также присутствующая в острой фазе). МРТ головного мозга обычно показывает Т2-гиперинтенсивный сигнал от базальных ганглиев (хвостатых ядер и скорлупы) (рис. 4 [A и B при BTRE]) в случае синдрома Лея и от срединных структур таламуса и околоводопроводного серого вещества в случае энцефалопатии Вернике [14]. При BTRE в острых фазах заболевания было описано поражение коры головного мозга с рассеянными гиперинтенсивными по Т2 очагами [15,16]. Несмотря на то, что тиамин является основой лечения обоих заболеваний, при BTRE также рекомендуется принимать добавки с биотином, которые, вероятно, повышают экспрессию SLCA19A3 посредством биотинилирования гистонов [17]. Кетогенная диета назначается при PDHAD, поскольку она увеличивает выработку печенью кетоновых тел, альтернативного глюкозе энергетического субстрата для образования ацетил-КоА в центральной нервной системе [18].
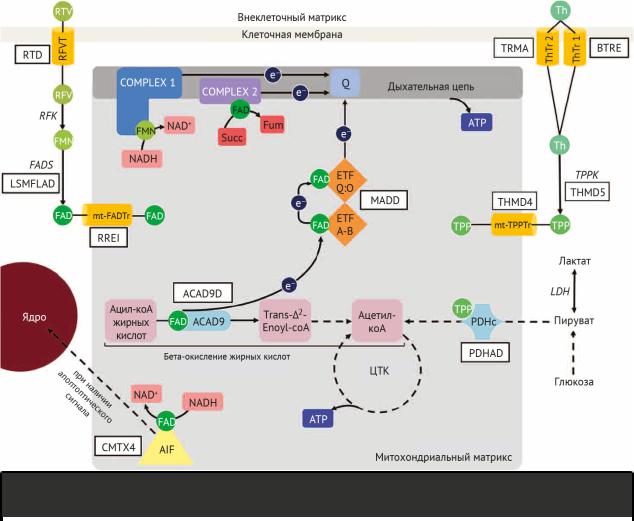
Рисунок 3 | Тиамин- и биотин-связанные метаболические пути

Заболевания, вызванные нарушением определенных реакций, обозначены белыми прямоугольниками. Биотин-тиамин-зависимая энцефалопатия (BTRE) является следствием нарушения внутриклеточного транспорта тиамина, вызванного дефектом переносчика тиамина-2 (ThTr-2), рецептора, который в основном экспрессируется в ЦНС; тиамин в своей активной форме, такой как тиаминпирофосфат, действует как кофактор пируватдегидрогеназного комплекса (PDHc), а также участвует в специфичных для ЦНС биохимических реакциях, таких как выработка ацетилхолина, поглощение серотонина и γ- аминомасляной кислоты. Дефицит пируватдегидрогеназы e1-α (PDHAD) вызван нарушением работы PDHc, который отвечает за превращение пирувата в ацетилкоА и, в частности, его субъединицу E1α, которая действует путем декарбоксилирования пирувата; этот дефект нарушает окисление глюкозы в митохондриальном цикле трикарбоновых кислот, вызывая снижение выработки аденозинтрифосфата (АТФ) и усиление анаэробного гликолиза, что приводит к избытку пировиноградной и молочной кислот (при нормальном соотношении лактат: пируват). Дефицит переносчика рибофлавина (RTD) — это нарушение внутриклеточного транспорта рибофлавина, витамина, важного для производства флавинмононуклеотида (FMN) и флавинадениндинуклеотида (FAD), связанных с транспортом электронов на митохондриальном уровне (дыхательная цепь и транспорт из бета-окисление жирных кислот в дыхательной цепи). В это вовлечены два рецептора: транспортер рибофлавина 2 и 3 (RFVT2 и RFVT3). Множественный дефицит ацил-коА-дегидрогеназы (MADD) является результатом нарушения транспорта электронов в дыхательную цепь в результате β-окисления жирных кислот. В этом участвуют два FAD-зависимых белка: флавопротеины электронного переноса (ETF), состоящие из 2 субъединиц, A и B, получающих электроны от фермента ацил-coA дегидрогеназы 9 (ACAD9); и электронтранспортная флавопротеин убихинон-оксидоредуктаза (ETFQ: O), которая принимает электроны от ETF и передает их коэнзиму Q. Фермент ACAD9 - это дегидрогеназа, участвующая в первом этапе митохондриального β-окисления жирных кислот и сборке первого комплекса дыхательной цепи. Миопатия накопления липидов вследствие дефицита синтетазы FAD (LSMFLAD) вызвана нарушением синтеза FAD из FMN (полученного из рибофлавина). Непереносимость физических нагрузок, связана с дефектом митохондриального транспортера FAD. Х-связанная болезнь Шарко — Мари — Тута 4 типа (CMTX4) является результатом нарушения функции фактора, индуцированного апоптозом (AIF), FAD-зависимой NADH-оксидазы, которая также действует как регулятор апоптоза, мигрируя в ядро и модулирование транскрипции ДНК. ACAD9D указывает на дефицит ацил-коА-дегидрогеназы 9; е-, электрон; FAD, синтетаза ФАД; Фум, фумарат; ЛДГ, ген лактатдегидрогеназы; mt-FADTr, митохондриальный переносчик флавинадениндинуклеотидов; mt-TPPTr, митохондриальный переносчик тиаминпирофосфата; NAD+, никотинамидадениндинуклеотид; NADН, никотинамидадениндинуклеотид и водород;
Q, убихинон; RFV, рибофлавин; RFVT, переносчик рибофлавина; RFK, рибофлавинкиназа; Succ, сукцинат; THMD4, чувствительная к тиамину двусторонняя дегенерация полосатого тела и полинейропатия; THMD5, энцефалопатия из-за недостаточности тиаминпирофосфокиназы; ThTr-1, переносчик тиамина-1; TPP, пирофосфат тиамина, активная форма тиамина; ТППК, тиаминпирофосфокиназа; TRMA, тиамин-чувствительная мегалобластная анемия.
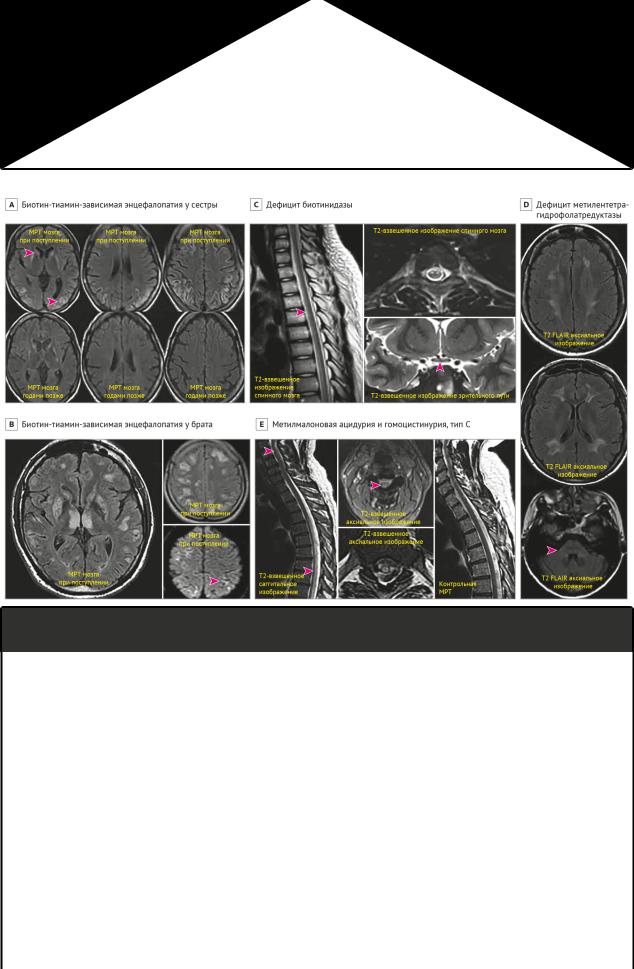
Рисунок 4 | МРТ-характеристики витамин-связанных заболеваний взрослых
А и В, биотин-тиамин-зависимая энцефалопатия среди сиблингов. A | МРТ головного мозга сестры демонстрирует двустороннюю
гиперинтенсивность головок хвостатых ядер (верхняя стрелка) и скорлупы, а также кортикальные и юкстакортикальные поражения белого вещества (нижняя стрелка) с двухсторонним гиперинтенсивным сигналом по Т2-FLAIR. Несколько лет спустя контрольная МРТ (изображения в нижнем ряду) показала почти полный регресс поражений. Вместо этого стала заметна серьезная диффузная атрофия мозга.
B | У брата была энцефалопатия по типу Вернике с симметричными измененными по T2-FLAIR сигналами от таламуса, наряду с гиперинтенсивностью базальных ганглиев и диффузными кортикальными и юкстакортикальными поражениями белого вещества. DWI-изображения демонстрируют некоторые тонкие очаги гиперинтенсивности, в основном внутри кортикальных поражений