

Лекции_1 / Лекция 2
.docЛекция 2. Механизм химической реакции
Понятие механизма реакции является одним из наиболее фундаментальных в современной химии. Знание механизма позволяет подобрать оптимальные условия получения необходимых продуктов, уменьшить нежелательные примеси, уменьшить энергетические затраты, улучшить экологические условия получения различных материалов. Знание механизма позволяет наиболее просто и естественно систематизировать большой по объему фактический материал, что существенно упрощает процесс обучения. Например, долгое время основным способом систематизации в органической химии был принцип изложения материала по классам органических соединений. Это приводило к резкому увеличению объемов учебников. Например, один из лучших учебников, используемых в университетах и химических вузах многих стран мира, написанный лауреатом Нобелевской премии по химии П. Карером имел объем около 80 печатных листов (1200 страниц). Использование классификации по механизму реакции позволило существенно уменьшить объем фактического материала и повысить уровень изложения.
Под механизмом реакции понимается последовательность элементарных превращений, приводящих взаимодействующие молекулы к продуктам реакции. В подавляющем большинстве случаев последовательность элементарных реакций не совпадает со стехиометрическими уравнениями. Пожалуй, одним из немногих исключений является реакция:
2NO2
![]() N2O4
N2O4
Хорошим примером, иллюстрирующим сложность возникающих проблем, является простейшая реакция — горение водорода. Стехиометрическое уравнение этой реакции выглядит так:
2Н2 + О2 = 2Н2О.
Однако хорошо известно, что это уравнение не имеет практически никакого отношения к тому, как на самом деле протекает химическое превращение. Если смешать водород с кислородом при комнатной температуре в темноте, то не удастся обнаружить никакой реакции. В то же время при повышении температуры или световом воздействии может произойти взрыв. Эта реакция исследовалась кинетиками в течение многих лет, так как она является простейшим примером открытых Н.Н. Семеновым (Семенов Н.Н., 1925) цепных разветвленных реакций, к которым относятся очень многие практически важные процессы горения.
На самом деле в смеси водорода с кислородом происходит большое число так называемых элементарных реакций.
В частности, установлено, что при достаточно низких давлениях основную роль играют следующие реакции:
-
Н2 + О2 → ОН + ОН – 18.6 ккал/моль — зарождение цепей,
-
ОН + Н2 → Н2О + Н + 14.7 ккал/моль — продолжение цепей,
-
Н + О2 → ОН + О – 16.6 ккал/моль — разветвление,
-
О + Н2 → ОН + Н – 1,9 ккал/моль — разветвление,
-
О, Н, ОН + стенка — гибель.
Полная схема реакции гораздо сложнее — она включает 24 реакции. Изучение таких сложных реакций, когда наряду с химическими реакциями надо учитывать диффузионные процессы, а также процессы тепло- и массопереноса, составляет предмет макрокинетики. Однако, как правило, определяющую роль в макрокинетическом процессе играют разыгрывающиеся на микроуровне элементарные процессы химического превращения при столкновениях молекул.
Строго говоря, полное исследование механизма реакции предполагает получение полной информации об изменении координат и энергий взаимодействующих на частицу в процессе реакции.
Молекулы состоят из атомов, занимающих определенные положения относительно друг друга. Это расположение атомов является, как правило, положением стабильного равновесия: небольшие смещения любого из атомов приводят к появлению сил, возвращающих их в равновесные положения. Спектральные и диффракционные методы часто позволяют определять расстояния между атомами в молекулах (длины связей) с точностью до ±1 нм, а значения углов между связями — в пределах ±0,5°.
Если смещения одного или нескольких атомов в одной или нескольких молекулах сравнительно велики, исходная структура молекул может не сохраниться: вместо этого атомы займут новые стабильные конфигурации в новых молекулах, т. е. произойдет химическая реакция. Для осуществления достаточно больших смещений и, следовательно, химической реакции необходима энергия, превышающая среднюю энергию, которой обладают молекулы при данной температуре. При любой определенной температуре столкновения между молекулами будут приводить к молекулам с различной энергией. При этом некоторые молекулы могут приобретать энергию, достаточную реакции. Чем выше температура, тем выше средняя молекул и тем большее число молекул иметь энергию, большую критической, необходимую для протекания реакции. Молекулы могут приобретать энергию и другими путями, наиболее важный источник энергий — видимый или ультрафиолетовый свет, поглощение которого приводит к возбуждению электронных уровней молекулы. Для того чтобы полностью понять механизм органической реакции, необходимо знать точное положение атомов в реагирующих молекулах как функцию от времени в процессе их превращения в продукты реакции через любые возможные интермедиаты.
Это - цель, которая никогда не может быть достигнута в полном объеме. Спектральные и диффракционные методы, с помощью которых определяется химическая структура молекулы, мало приспособлены для наблюдения за изменениями структуры, которые происходят в течение химических реакций за время 10-13 — 10-14 с, так как это время сравнимо со временем, необходимым для молекулярного колебания или столкновения молекул. Следовательно, наше знание механизма реакций должно основываться на косвенных данных.
Можно получить данные, которые опровергнут тот или иной предлагаемый механизм реакции, однако имеющиеся данные полностью доказывают правильность предложенного механизма. Учитывая это, механизм реакции считается установленным, когда широкий ряд данных свидетельствует в его пользу. Однако с течением времени могут появиться новые факты, которые заставят видоизменить или отбросить этот механизм.
В настоящее время для изучения механизмов химических реакций используется большое число экспериментальных и теоретических методов исследования. С некоторыми их этих методов мы познакомимся на следующих лекциях. Отметим, что в последние десятилетия ценную информацию о механизмах сложных многостадийных реакций удалось получить на основе современных квантово-химических методов.
Во многих случаях результаты теоретического исследования хорошо согласуются с экспериментальными данными и позволяют получить важный дополнительный материал о механизмах реакций, а также о влиянии заместителей на величину энергии активации.
Реакция элиминирования HNO2 через плоское пятичленное переходное состояние является одним из наиболее изученных теоретически и экспериментально процессов термического распада нитросоединений. Впервые механизм этой реакции был теоретически изучен около 30 лет назад с использованием метода MINDO/3. Полученные результаты позволили сделать важный для понимания основных закономерностей влияния заместителей на энтальпию (энергию) активации реакции вывод о том, что переходное состояние, вопреки принятым ранее представлениям, является неполярным, т.е. дипольный момент ПС меньше, чем у исходных молекул (нитроэтана, 1-нитропропана и ряда других нитроалканов, не содержащих атомов галогенов). В дальнейшем этот вывод был подтвержден расчетами с использованием неэмпирических и DFT-методов, которые позволили добиться хорошего согласия экспериментальных и расчетных значений энергии активации. Теоретическое изучение реакции элиминирования HNO2 особенно подробно проводилось для нитроэтана (табл. 2). Полученные с использованием ряда неэмпирических и DFT-методов значения энтальпии активации реакции хорошо согласуются с экспериментальными значениями энергии активации газофазного распада нитроэтана (188.3 кДж·моль-1).
Результаты неэмпирических и полуэмпирических методов дают несколько различающуюся картину структуры ПС и механизма реакции. По данным методов MINDO/3 и РМЗ, в ПС наиболее сильно увеличивается длина связи С–Н, образованная участвующим в реакции атомом водорода. Все остальные использованные методы предсказывают, что наиболее сильно в ПС изменяется длина связи С–N, которая практически уже разорвана. Учитывая, что полуэмпирические методы существенно завышают энтальпию (энергию) активации реакции, а неэмпирические и DFT-методы дают в целом хорошо согласующиеся с результатами эксперимента оценки, можно полагать, что их предсказания структуры ПС являются более надежными. Поскольку метод B3LYP с различными базисами дает достаточно надежные результаты, дальнейшее изучение влияния заместителей в работах проводилось с использованием метода B3LYP/6-31G(d). Наиболее важные из полученных данных представлены в табл. 2.
Анализируя основные изменения геометрических параметров реакционного центра в переходном состоянии по сравнению с исходными молекулами (см. табл. 1-2), можно объяснить изменение энтальпии активации реакции в ряду нитроалканов. Так, для нитроалканов нормального строения уменьшение энтальпии активации можно связать с уменьшением в ПС величин r(C–H) и r(N–О). Например, эти величины в реакциях 1-нитропропана и 1-нитробутана меньше, чем для нитроэтана, на 2.8 и 0.4 пм соответственно. С учетом различий в прочности связей эти изменения перекрывают относительно более сильное растяжение связи С–N для указанных соединений, поэтому энтальпии активации для них меньше, чем для нитроэтана. Интересно, что как геометрические параметры исходных молекул и ПС, так и барьеры реакций элиминирования HNO2 из 1-нитропропана, 1-нитробутана и 1-нитропентана практически совпадают. Отметим, что для многих изученных нитроалканов изменения энтальпии активации и энергии диссоциации связи С–N происходят согласованно (рис. 1).
Если в реакции участвуют группы NO2 при вторичном или третичном атоме углерода, уменьшение энтальпии активации происходит монотонно в ряду соединений с увеличением длины формирующейся в ПС двойной связи С=С. Понятно, что чем больше длина этой связи, тем меньшим энергетическим затратам соответствует перестройка одинарной связи С–С в двойную в ПС. Согласие расчетных и экспериментальных значений энергий активации можно признать достаточно хорошим. Средняя погрешность расчета для реакций мононитроалканов составляет 5.6 кДж-моль-1.
Таблица 1. Основные геометрические параметры переходного состояния и энтальпии активации реакции элиминирования HNO2 из нитроэтана по данным различных квантово-химических методов.


Для динитроалканов точность экспериментальных значений энергий активации газофазного элиминирования HNO2, пересчитанных из данных, которые получены для реакции присоединения азотистой кислоты к алкенам, значительно ниже. Однако и в этом случае в целом достигается удовлетворительное согласие расчетных и экспериментальных величин энергии активации.
Таблица 2. Геометрические параметры переходного состояния и энергии активации реакции элиминирования HNO2 из некоторых нитроалканов (метод B3LYP/6-31G(d), Т = 600 К) (нумерацию атомов см. в табл. 1).

Значительный интерес представляют теоретические оценки барьеров реакции элиминирования HNO2 из 1,1-динитроэтана и 1,1-динитропропана. Расчет предсказывает в этом случае небольшое (на 13.5 и 13.9 кДж-моль-1) уменьшение энтальпии активации реакции по сравнению с нитроэтаном и нитропропаном соответственно. Хотя энергия активации радикального распада и величина D(C–N) для этих соединений уменьшаются сильнее (на 30-35 кДжмоль-1), все же при проведении экспериментальных исследований нельзя полностью исключать возможный вклад распада по механизму элиминирования в суммарную константу скорости. При этом следует иметь в виду, что по сравнению с мононитросоединениями экспериментальное изучение гем-динитроалканов проводилось при существенно более низких температурах.
Определенные успехи были достигнуты и при расчете предэкспоненциального множителя реакции. Впервые теоретическая оценка Л-фактора реакции газофазного элиминирования HNO2 для нитроэтана, 1-нитропропана, 1-нитробутана была проведена полуэмпирическим методом MINDO/3 с учетом заторможенного вращения функциональных групп, непосредственно примыкающих к реакционному центру, на основе модели симметричного волчка. Результаты расчета в целом достаточно хорошо передавали как тенденции изменения в ряду соединений (уменьшение lgA при переходе от нитроэтана к 1-нитробутану), так и абсолютные значения предэкспоненциального множителя. При этом расчетные оценки величины lgA систематически превышают результаты эксперимента на 0.25-0.3. Анализ полученных данных показал, что вращательные составляющие статистической суммы практически не дают вклада в изменение lgA, что связано с незначительным изменением моментов инерции молекул в процессе перехода из основного состояния в переходное. Статистическая сумма внутреннего вращения в ПС во всех соединениях меньше, чем в основном состоянии, причем практически на одну и ту же величину. Из этих данных был сделан вывод, что в ПС происходит «замораживание» внутреннего вращения группы NO2. Основной вклад в изменение Л-фактора реакции вносит колебательная составляющая, во всех случаях ее значение в ПС больше, чем в основном состоянии. Известно, что метод MINDO/3 дает большие ошибки при расчете частот колебаний. Вместе с тем частотные вклады в колебательную статистическую сумму правильно отражают реальную тенденцию изменения А-фактора реакции в ряду нитроалканов.
В настоящее время получены достаточно подробные расчетные данные по частотам колебаний и барьерам вращения функциональных групп 15 в молекулах нитроалканов, которые хорошо согласуются с имеющимися (к сожалению, малочисленными) результатами эксперимента. Это позволило более подробно рассмотреть особенности влияния строения молекул на изменение в ряду нитроалканов предэкспоненциального множителя реакции газофазного элиминирования HNO2.
Проведенное с использованием гибридного DFT-метода B3LYP исследование показало, что неплохие результаты для всех экспериментально изученных нитроалканов могут быть получены на основе наиболее простой модели — полностью заторможенного вращения функциональных групп в исходных молекулах и ПС. С учетом того, что в ПС часть вращений «заморожена», данная модель должна давать завышенные оценки Л-фактора. Результаты расчета подтверждают, что для большинства реакций действительно наблюдается подобная тенденция, однако согласие расчетных и экспериментальных оценок в целом следует признать достаточно хорошим. Исключение составляют расчетные значения А-фактора реакции элиминирования HNO2 для соединений с группами NO2 у вторичных и третичных атомов углерода. Установлено, что для всех изученных экспериментально нитроалканов хорошо согласующиеся с экспериментом оценки могут быть получены при использовании наиболее общей модели – заторможенного асимметричного вращения всех функциональных групп реакционного центра. Хорошее согласие расчетных и экспериментальных значений lgА проиллюстрировано на рис. 2.
Приведенные данные показывают перспективность использования современных квантово-химических методов для оценки А-фактора реакции газофазного элиминирования HNO2. Вместе с тем эта важная и интересная проблема нуждается в дополнительном изучении.

Рис. 2. Корреляция между значениями ΔH# и D(C–N) для нитроалканов (коэффициент корреляции 0.954).
1 — (CH3)3CNO2, 2 — (CH3)2C(NO2)CH2CH3, 3 — (CH3)2C(NO2)CH(CH3)2, 4 — CH3CH(NO2)CH2C3H7, 5 — CH3CH(NO2)CH3, 6 — CH3CH(NO2)CH2CH3, 7 — CH2(NO2)CH2CH(CH3)2, 8 — C5H,2CH2NO2, 9 — C2H5CH2NO2, 10 — C3H7CH2NO2, 11 — CH2(NO2)CH(CH3)2, 12 — CH3CH2NO2.
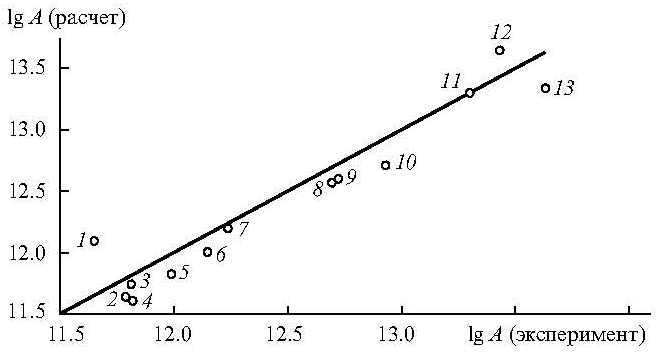
Рис. 3. Корреляция между расчетными (с учетом асимметричного внутреннего вращения относительно всех связей, примыкающих к реакционному центру) и экспериментальными значениями lgА (с-1) для реакции элиминирования HNCh из нитроалканов (Т = 600К) (коэффициент корреляции 0.958). 1 — CH2(NO2)CH(CH3)2, 2 — C3H7CH2NO2, 3 — C5H12CH2NO2, 4 — C4H9CH2NO2, 5 — CH2(NO2)CH2CH(CH3)2, 6 — C2H5CH2NO2, 7 — CH3CH2NO2, 8 — CH3CH(NO2)CH2CH3, 9 — CH3CH(NO2)CH3, 10 — CH3CH(NO2)CH2C3H7, 11 — (CH3)2C(NO2)CH2CH3, 12 — (CH3)2C(NO2)CH(CH3)2, 13 — (CH3)3CNO2.
В дальнейшем мы рассмотрим некоторые особенности использования современных квантово-химических методов для изучения механизмов химических реакций более подробно. Отметим, что в настоящее время квантово-химические методы стали важнейшим инструментом изучения механизмов химических реакций.