

ОБЩИЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ
МЕТОДЫ АНАЛИЗА
ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
1. ХРОМАТОГРАФИЯ (ОФС 42-0092-09)
Хроматографией называется физико-химический метод разделения смесей, в котором разделяемые компоненты распределены между двумя ф а-
зами. Одна из этих фаз (стационарная фаза) неподвижна, а другая (подвиж-
ная фаза) постоянно движется в определенном направлении .
По свойствам подвижной и неподвижной фаз хроматографические ме-
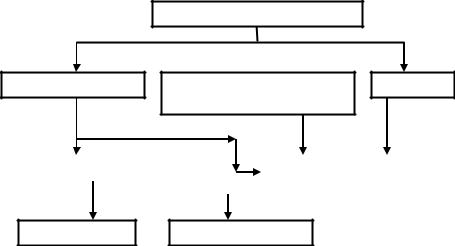
тоды можно разделить на следующие типы, показанные на схеме (рис. 1.1).
ХРОМАТОГРАФИЯ
ЖИДКОСТНАЯ  СВЕРХКРИТИЧЕСКАЯ
СВЕРХКРИТИЧЕСКАЯ  ГАЗОВАЯ ФЛЮИДНАЯ
ГАЗОВАЯ ФЛЮИДНАЯ
ПЛАНАРНАЯ |
|
|
КОЛОНОЧНАЯ |
|
|
|
|
|
|
|
|
|
|
|
БУМАЖНАЯ ТОНКОСЛОЙНАЯ
Рис. 1.1. Типы хроматографии Результат хроматографического разделения представляется в виде хро-
матограммы.
Хроматограмма и хроматографичеcкие параметры
Хроматограммой называют последовательность пятен, зон или пиков,
соответствующих компонентам исходной смеси после их хроматографиче-
ского разделения. Для любого типа хроматографического процесса (см.
рис. 1.1), в котором для регистрации результата используется детектор, хро-
матограмма представляет собой график зависимости сигнала детектора, про-
порционального концентрациям разделяемых компонентов, от времени (ко-
лоночная хроматография) или расстояния (планарная хроматография). В пла-
нарной хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или на ТСХ-пластинке (тонкослойная хроматография) последовательность пятен компонентов исходной (анализи-
руемой) смеси.
Схема типичной хроматограммы приведена на рис. 1.2. Хроматограмма состоит из пиков, разделенных базовой линией.
Базовая линия соответствует сигналу только от подвижной фазы.
Пик – кривая, в идеале приближающаяся к кривой гауссова распреде-
ления, которая описывает постепенное нарастание концентрации вещества и последующее ее уменьшение.
СИГНАЛ ДЕТЕКТОРА
Рис. 1.2. Хроматограмма и основные хроматографические параметры
1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося компонента; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)

ИНТЕРПРЕТАЦИЯ ХРОМАТОГРАФИЧЕСКИХ ДАННЫХ
ti – время удерживания компонента – соответствует времени появления максимума пика на хроматограмме, где i – индекс, соответствующий i-му компоненту разделяемой смеси. При точном воспроизведении параметров хроматографической системы время удерживания является величиной, по-
стоянной для данного вещества;
t0 – время удерживания несорбирующегося компонента;
tR = ti − t0 – приведенное (исправленное) время удерживания компонен-
та;
Rr – относительное время удерживания компонента 2 по компоненту 1
Rr t2 t1
t1 и t2 – времена удерживания двух компонентов, измеренные от момен-
та инжекции.
r – относительное удерживание компонента 2 по компоненту 1
rt2 t0 t1 t0
k = (ti – t0)/t0 – коэффициент емкости колонки по i компоненту;
W – ширина пика у основания (для пиков, описываемых гауссовым за-
коном распределения, W = 4 , см. рис. 1.2);
W0,5 – ширина пика на половине высоты (для пиков, описываемых гаус-
совым законом распределения, W0,5 = 2,35 , см. рис. 1.2);
R – разрешение между пиками двух компонентов смеси может быть
описано уравнениями: |
|
||
R |
2 (t2 t1 ) |
, |
(1) |
|
|||
|
W1 W2 |
|
|
или
R |
1,18 (t2 |
t1) |
, |
(2) |
|
|
(W0,5)1 (W0,5)2
При R ≥ 1,5 компоненты разделены полностью (до базовой линии).
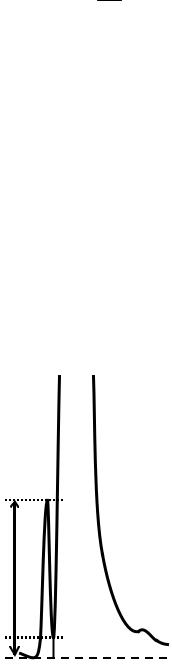
Коэффициент p/v (отношение «пик – впадина»)
p/v Hp , Hv
Данное соотношение используют для оценки эффективности хромато-
графической системы, если компоненты смеси разделяются не полностью
(рис. 1.3).
N – число теоретических тарелок, определяется следующими выраже-
ниями:
t |
i |
2 |
|
||||
N 16 |
|
, |
(3) |
||||
W |
|
||||||
|
i |
|
|
||||
или |
|
|
|
2 |
|
||
|
ti |
|
|
||||
N 5,54 |
|
|
|
|
. |
(4) |
|
|
|
|
|
||||
W |
|
|
|
||||
|
i; 0,5 |
|
|||||
Hp
Hv
Рис. 1.3. Хроматограмма не полностью разделяемых компонентов
Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии
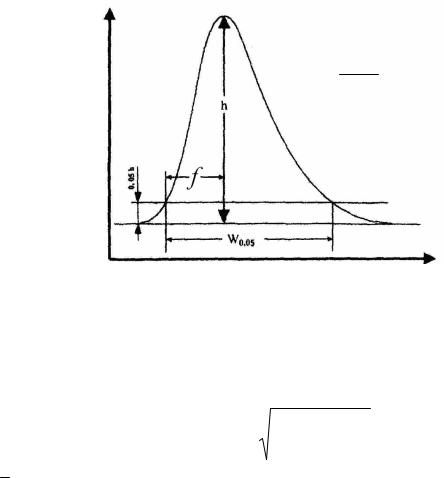
T – фактор асимметрии пика (хвостовой фактор) рассчитывают по
уравнению (см. рис. 1.4):
T |
W0,05 |
, |
(5) |
|
|||
|
2 f |
|
|
где: W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты.
При T = 1 пик симметричен.
Сигнал детектора
Время
T W0,05
2 f
Рис. 1.4. Схема расчета фактора асимметрии пика
RSD (%) – относительное стандартное отклонение, вычисляемое по
формуле:
|
100 |
|
n |
( X i |
|
|
)2 |
|
|
|||
|
|
X |
|
|||||||||
RSD |
|
|
|
|
|
|
|
|
, |
(6) |
||
|
|
|
|
|
|
|||||||
|
|
X |
|
i 1 |
n 1 |
|
||||||
где: X – среднее результатов определений;
Xi – результаты единичных определений;
n – число определений.
Для оценки пригодности хроматографической системы в колоночной
хроматографии обычно используют следующие параметры: R; p/v; N; T; RSD,
–как по отдельности, так и в различных сочетаниях.
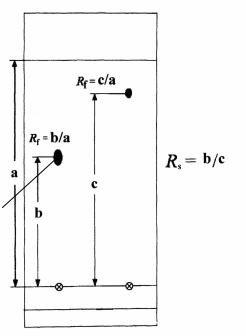
Впланарной хроматографии аналогом времени удерживания является фактор удерживания:
Rf = a/b, |
(7) |
где: a – расстояние от точки нанесения пробы до центра пятна; b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют усло-
вия хроматографирования. Оценкой хроматографической подвижности, ме-
нее чувствительной к влиянию отклонений в условиях проведения экспери-
мента, является величина Rs, представляющая собой отношение величины Rf
одного вещества к величине Rf другого, принятого за стандарт. Величины Rf
и Rs используют для идентификации веществ. Обычно выбор стандарта осу-
ществляется так, чтобы значение Rs анализируемого вещества было в преде-
лах от 0,5 до 2,0. Схема определения этих величин приведена на рис. 1.5.
ЛИНИЯ ФРОНТА
 СТАНДАРТ
СТАНДАРТ
ОПРЕДЕЛЯЕМОЕ
ВЕЩЕСТВО
ЛИНИЯ СТАРТА УРОВЕНЬ ЭЛЮЕНТА
Рис. 1.5. Схема определения значений Rf и Rs
 – место нанесения образца на линию старта
– место нанесения образца на линию старта
Данные планарной хроматографии могут быть представлены в виде
денситограмм.
Расчет содержания определяемого компонента
Существуют 3 основных метода расчета концентрации анализируемого компонента по хроматографическим данным.
1. Метод нормирования (метод внутренней нормализации, нормировки).
Применение данного метода основано на предположении, что на хрома-
тограмме зарегистрированы все компоненты, входящие в состав анализируе-
мой смеси, и что доля площади (высоты) каждого пика от суммы площадей
(высот) всех пиков соответствует содержанию компонента в массовых про-
центах. Содержание каждого компонента в процентах может быть вычислено по формулам:
|
|
X i |
Si |
100 |
|
||||
|
|
|
|
|
|
, |
(8) |
||
|
|
|
|
n |
|
||||
|
|
|
|
Si |
|
||||
|
|
|
|
i 1 |
|
||||
|
|
или |
|
|
|
|
|
||
|
|
X i |
Si |
100 |
|
||||
|
|
|
|
|
, |
(8а) |
|||
|
|
|
|
|
|||||
|
|
|
|
|
|
S |
|
||
где: Si |
– |
площадь (высота) i-го пика; |
|
||||||
n |
|
|
|
|
|
|
|
|
|
Si |
– |
сумма площадей (высот) всех пиков на хроматограмме; |
|
||||||
i 1 |
|
|
|
|
|
|
|
|
|
S |
– |
площадь (высота) основного пика. |
|
||||||
Если чувствительность детектора различна по отношению к каждому из компонентов, то вводят поправочные коэффициенты ki. Выражение (8)
приобретает вид:
X i |
k S |
100 |
|
|
i i |
|
, |
(9) |
|
n |
|
|||
ki Si
i 1
Поправочные коэффициенты рассчитывают относительно основного компонента анализируемой смеси или другого стандартного вещества по формуле:
ki |
|
Ci S0 |
, |
(10) |
|
C0 Si |
|||||
|
|
|
|
где: Сi и С0 – концентрация i-ого компонента и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-ого компонента и стандартного вещества соответственно.
2. Метод внешнего стандарта. Готовят раствор стандартного образца определяемого компонента с концентрацией, близкой к предполагаемой ко н-
центрации этого компонента в испытуемой пробе. Последовательно хромато-
графируют стандартный, испытуемый и снова стандартный растворы (каж-
дый не менее 3-х раз). Находят средние значения площадей пиков на хрома-
тограммах и рассчитывают концентрацию определяемого компонента в ис-
пытуемом растворе по уравнению:
|
|
C |
S C0 |
, |
(11) |
|
|
|
|||
|
|
|
S0 |
|
|
где: S и S0 |
– |
средние значения площадей (высот) пиков на хроматограммах |
|||
|
|
испытуемого и стандартного растворов соответственно; |
|
||
С и С0 |
– |
концентрации определяемого и стандартного растворов со- |
|||
|
|
ответственно. |
|
||
Для проведения анализа методом внешнего стандарта необходимо пре д-
варительно убедиться, что в используемом диапазоне концентраций площадь пика определяемого компонента линейно зависит от его концентрации.
3. Метод внутреннего стандарта. Метод внутреннего стандарта ос-
нован на том, что в анализируемую смесь вводят определенное количество стандартного вещества (внутренний стандарт). Это вещество должно быть химически инертным, отсутствовать в анализируемом образце и полностью отделяться от других компонентов смеси. Время его удерживания должно
быть близким к ti определяемых компонентов, концентрация должна быть
близка к концентрациям определяемых компонентов, пик симметричным.
В испытуемый и стандартный растворы вводят равные количества
внутреннего стандарта, хроматографируют и определяют отношения площа-
дей (высот) пиков определяемого компонента к площади (высоте) пика внут-
реннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого компонента (Х) рассчитывают по фор-
муле: |
|
|
|
|
|
X |
B C0 |
, |
(12) |
|
|
|||
где: B=S/S0 |
|
B0 |
|
|
– отношение площади (высоты) пика определяемого компо- |
||||
|
нента к площади (высоте) пика внутреннего стандарта в |
|||
|
испытуемом растворе; |
|
||
B0=S/S0 |
– отношение площади (высоты) пика определяемого компо- |
|||
|
нента к площади (высоте) пика внутреннего стандарта в |
|||
|
стандартном растворе; |
|
||
С0 |
– концентрация стандартного раствора. |
|
||