

Книги по МРТ КТ на английском языке / Advanced Imaging of the Abdomen - Jovitas Skucas
.pdf
 B
B


633
KIDNEYS AND URETERS
Multiple Myeloma
Renal histology in patients with myeloma nephropathy revealed 14% with tubulointerstitial nephritis, 11% with amyloidosis, 7% with acute tubular necrosis, and 4% each with nodular glomerulosclerosis and plasma cell infiltration (83); at times renal biopsy pro-vides the first clue to a diagnosis of myeloma by identifying myeloma cast nephropathy.
Tubular precipitation of Bence-Jones proteins |
|
|
is a cause of renal failure in multiple myeloma |
|
|
patients. Haphazard IV contrast injection is a |
|
|
cause of such precipitation, but with preplan- |
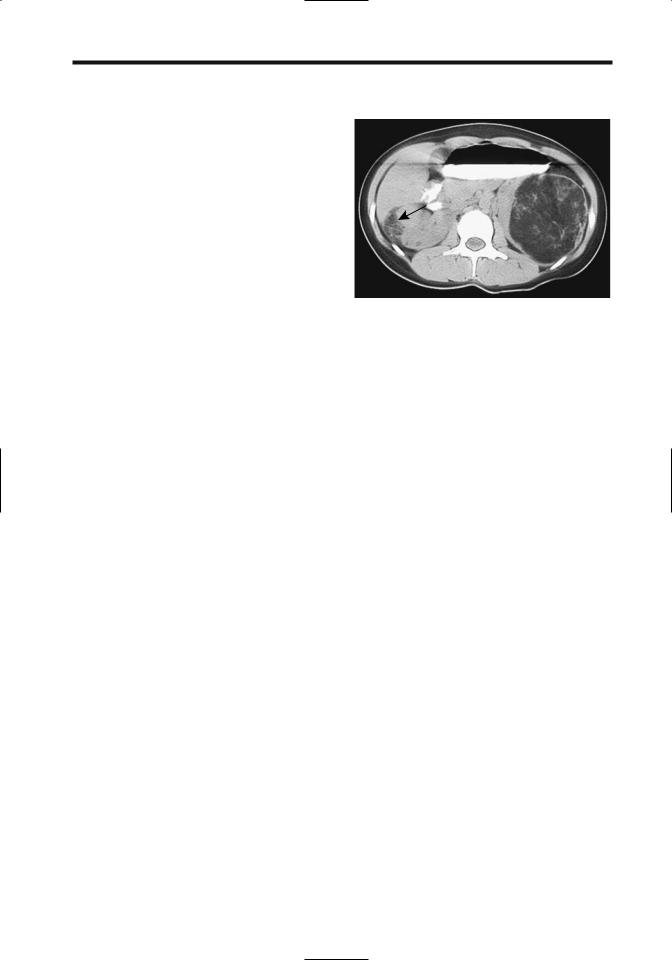
Figure 10.27. Renal angiomyolipomas in a 17-year-old with |
|
ning and adequate hydration a number of these |
||
tuberous sclerosis. The left fat-containing tumor is obvious. A |
||
patients have undergone contrast studies |
smaller angiomyolipoma is also present in the right kidney |
|
without complication. |
(arrow). (Courtesy of Luann Teschmacher, M.D., University of |
|
Resorption of calcium from bone and the |
Rochester.) |
|
resultant hypercalcemia lead to nephrocalci- |
|
|
nosis. The kidneys enlarge, and the collecting |
|
|
systems are compressed. These patients are also |
|
|
prone to developing uric acid calculi. |
oleiomyomatosis, and tumors at other sites, but |
|
Unexplained renal failure in a patient even |
in general extrarenal angiomyolipomas are rare. |
|
with absent skeletal lesions can still be due to |
These tumors develop in the renal capsule, |
|
multiple myeloma. |
cortex, and medulla. Histologically they are |
|
|
composed of haphazardly arranged blood |
|
Mesenchymal Neoplasms |
vessels, disorganized smooth muscle fibers, and |
|
Angiomyolipoma |
varying amounts of fat. Some contain sheets of |
|
epithelioid cells,suggesting a malignancy. Those |
||
Clinical |
without fat mimic a leiomyoma. Histologically, |
|
no sarcomatous transformation should be |
||
|
||
Classification of angiomyolipomas has evolved |
evident in either the leiomyomatous or |
|
from their being considered hamartomas to |
lipomatous components. |
|
benign neoplasms, although a rare one does |
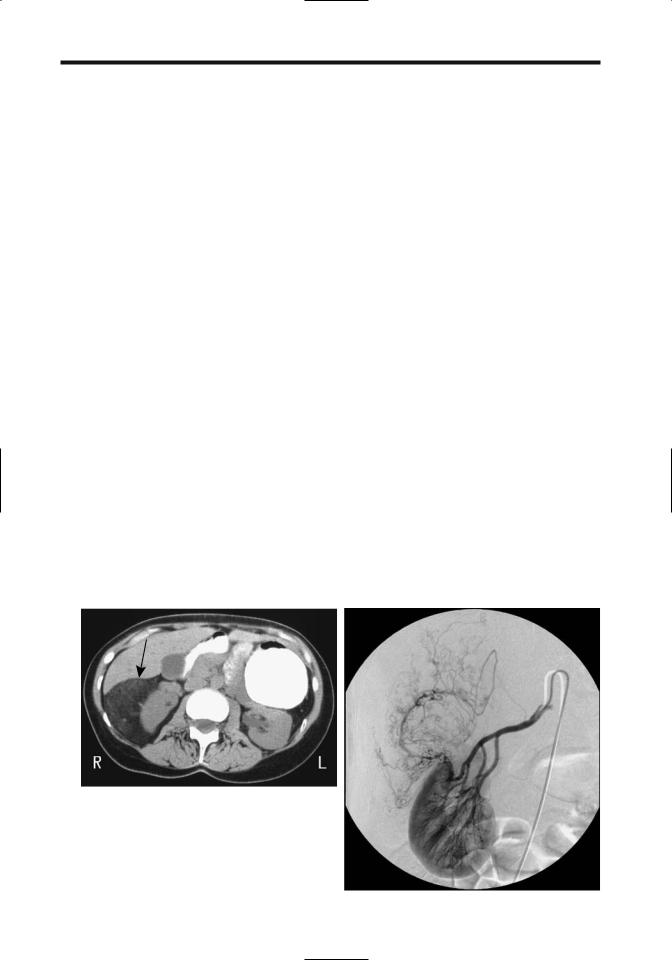
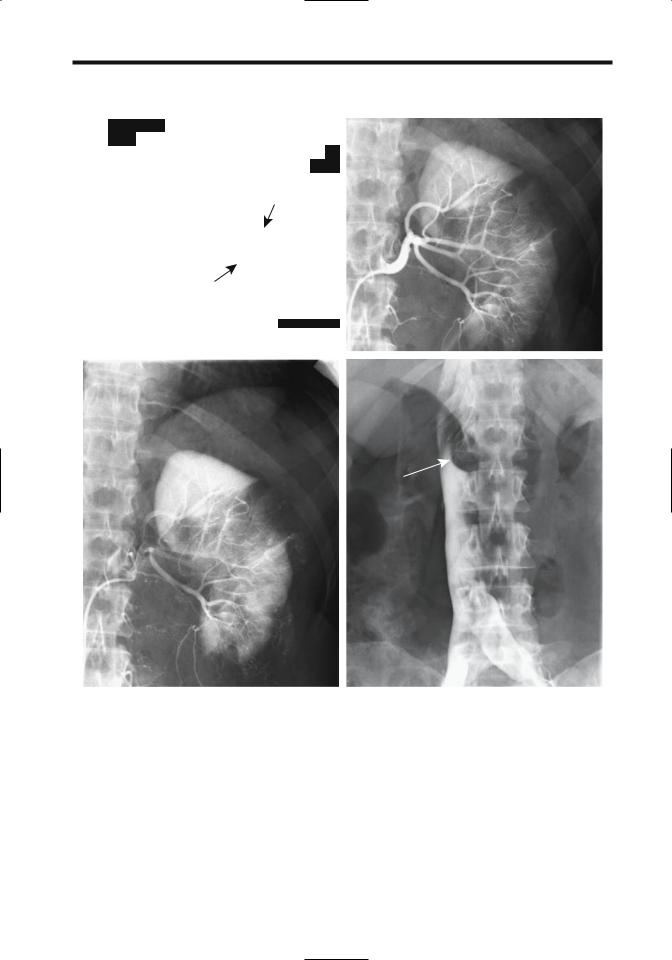
Serial CT and US provide a measure of renal |
|
undergo sarcomatous transformation and a |
angiomyolipoma growth rates. The mean |
|
propensity to metastasize. Even the usually |
growth rate of isolated tumors is about 5% per |
|
benign variety tends to be locally aggressive and |
year, being considerably more in those with |
|
produces considerable mischief. In some studies |
tuberous sclerosis. No correlation exists |
|
20% to 50% are detected in tuberous sclerosis |
between the amount of fat in a tumor and its |
|
patients, where they tend to be multiple and |
growth rate. Growth tends to be unpredictable. |
|
bilateral. Angiomyolipomas are associated to a |
Some tumors are stable for years and then |
|
lesser degree with von Recklinghausen’s disease, |
undergo a fast increase in size, followed by |
|
von Hippel-Lindau disease, and adult polycystic |
another period of quiescence. Rapid growth |
|
disease, and are found in about half of patients |
occasionally occurs in pregnancy, suggesting |
|
with pulmonary lymphangiomyomatosis. In |
that hormones play a role in their growth. An |
|
general, a finding of multiple angiomyolipomas |
occasional angiomyolipoma is huge at initial |
|
should suggest tuberous sclerosis (Fig. 10.27). |
presentation. |
|
In tuberous sclerosis, angiomyolipomas occur |
Spontaneous, solitary tumors are most |
|
earlier in life than sporadic ones, and most |
common in middle-aged women and are rare in |
|
are discovered incidentally. An occasional |
children. Pain, a palpable tumor, or hematuria is |
|
tuberous sclerosis patient develops bilateral |
a common presentation, although many of these |
|
renal angiomyolipomas,pulmonary lymphangi- |
tumors are asymptomatic. |
 B
B

