

- •Pineal Parenchymal Tumors
- •Germ Cell Tumors
- •Selected References
- •Medulloblastoma
- •Selected References
- •Anatomy of the Cranial Meninges
- •Meningomas
- •Primary Melanocytic Lesions
- •Other Related Neoplasms
- •Selected References
- •Cranial Nerve Anatomy
- •Schwannomas
- •Neurofibromas
- •Selected References
- •Histiocytic Tumors
- •Selected References
- •Sellar Region Anatomy
- •Normal Imaging Variants
- •Congenital Lesions
- •Neoplasms
- •Miscellaneous Lesions
- •Selected References
- •Intracranial Pseudotumors
- •Selected References
- •Metastatic Lesions
- •Paraneoplastic Syndromes
- •Selected References
- •Scalp Cysts
- •Extraaxial Cysts
- •Parenchymal Cysts
- •Intraventricular Cysts
- •Selected References
- •Anatomy and Physiology of the Basal Ganglia and Thalami
- •Selected References
- •Alcohol and Related Disorders
- •Opioids and Derivatives
- •Inhaled Gases and Toxins
- •Selected References
- •Selected References
- •Hypertensive Encephalopathies
- •Glucose Disorders
- •Thyroid Disorders
- •Seizures and Related Disorders
- •Miscellaneous Disorders
- •Selected References
- •The Normal Aging Brain
- •Dementias
- •Degenerative Disorders
- •Selected References
- •Normal Variants
- •Hydrocephalus
- •CSF Leaks and Sequelae
- •Selected References
- •Cerebral Hemisphere Formation
- •Imaging Approach to Brain Malformations
- •Posterior Fossa Anatomy
- •Chiari Malformations
- •Hindbrain Malformations
- •Selected References
- •Commissural Anomalies
- •Malformations Secondary to Abnormal Postmigrational Development
- •Selected References
- •Anencephaly
- •Holoprosencephaly
- •Holoprosencephaly Variants
- •Related Midline Disorders
- •Holoprosencephaly Mimics
- •Selected References
- •Selected References
- •Selected References
- •Cephaloceles
- •Craniosynostoses
- •Meningeal Anomalies
- •Selected References
- •Index

Acquired Metabolic and Systemic Disorders
appear similar, and both disorders can cause hemorrhagic microangiopathy. The microbleeds of CAA are more often peripheral (e.g., cortex, leptomeninges) and rarely affect the brainstem or cerebellum. Hypertensive microhemorrhages are most common in the basal ganglia and frequently can be identified in the pons and cerebellar hemispheres.
CHRONIC HYPERTENSIVE ENCEPHALOPATHY
Pathology
•Microvasculopathy
○Arteriolosclerosis, lipohyalinosis
○Myelin pallor, lacunar infarcts
○Microbleeds (cerebellum, basal ganglia/thalami > cortex)
Clinical Issues
•Metabolic syndrome, headaches
•Can have "acute-on-chronic" HTN with encephalopathy
Imaging
•Diffuse patchy and/or confluent WM lesions
•Microbleeds on T2* (basal ganglia, cerebellum)
Differential Diagnosis
•Amyloid angiopathy (cortex > basal ganglia, cerebellum)
•CADASIL (younger patients, anterior temporal/external capsule WM lesions)
Cerebral autosomal-dominant arteriopathy without subcortical infarcts and leukoencephalopathy (CADASIL) can also mimic CHtnE. CADASIL typically presents in younger patients and causes multiple subcortical lacunar infarcts. Lesions in the anterior temporal lobes and external capsules are classic imaging findings of CADASIL.
Glucose Disorders
The brain is a glucose glutton, consuming more than half the body's total glucose. Because the brain does not store excess energy as glycogen, CNS function is highly dependent on a steady, continuous supply of blood glucose (see box below).
Blood glucose levels are tightly regulated and are normally maintained within a narrow physiologic range. Disorders of glucose metabolism—both hypoglycemia and hyperglycemia—can injure the CNS.
The neurologic manifestations of deranged glucose metabolism range from mild, reversible focal deficits to status epilepticus, coma, and death. Because the clinical and imaging manifestations differ in neonates from those of older children and adults, hypoglycemia in these two age groups is discussed separately.
The vast majority of hypoglycemia cases are acquired. A few inherited syndromes present with infantile hyperinsulinemic hypoglycemia as a secondary manifestation of systemic disease. The effects of hypoglycemia on the infant brain are
1029
identical, regardless of etiology, so they are discussed in this chapter.
Following the discussion of hypoglycemia, we turn our attention to hyperglycemia-associated disorders that affect the CNS.
GLUCOSE AND THE BRAIN
Normal Physiology
•Brain is a "glucose glutton"
○Utilizes 100-150 g/day
•Glucose must be actively transported across bloodbrain barrier
○Glucose transport protein (GLUT-1)
•Glucose metabolism
○Aerobic oxidation (20% of total body O consumption)
○Intracellular glucose converted to pyruvate
○Then metabolized to ATP, phosphocreatine
•Glucose utilization linearly related to CBF
○GM ≈ 5x WM
○Mostly used for active ion transport, maintenance of membrane potentials
•Glucose homeostasis
○Blood glucose concentration dynamic, tightly regulated
○Brain monitors, "conducts" gut-CNS-endocrine axis
○Complex interactions maintain normal glycemia
Abnormal Physiology
•Too much or too little glucose both injure brain
•Hyperglycemic > > hypoglycemic disorders
Pediatric/Adult Hypoglycemic
Encephalopathy
Terminology
Hypoglycemia literally means low blood sugar and is caused by an imbalance between glucose supply and glucose utilization. Acute hypoglycemic brain injury is called hypoglycemic encephalopathy.
Etiology
Childhood hypoglycemic encephalopathy is most commonly associated with type 1 diabetes mellitus. Rarely, hypoglycemia occurs as an inherited disorder (see below) or secondary to insulin-secreting tumors.
In its most common adult setting—advanced type 2 diabetes—hypoglycemia typically results from the interplay between absolute or relative insulin excess and compromised glucose counterregulation; insulin in and of itself is not neurotoxic. Most cases of adult hypoglycemia occur as a side effect of diabetes treatment with insulin and sulfonylureas.
Factors other than absolute blood glucose levels also affect the presence and extent of hypoglycemic brain injury, including the duration and severity of hypoglycemia, presence
Toxic, Metabolic, Degenerative, and CSF Disorders
1030
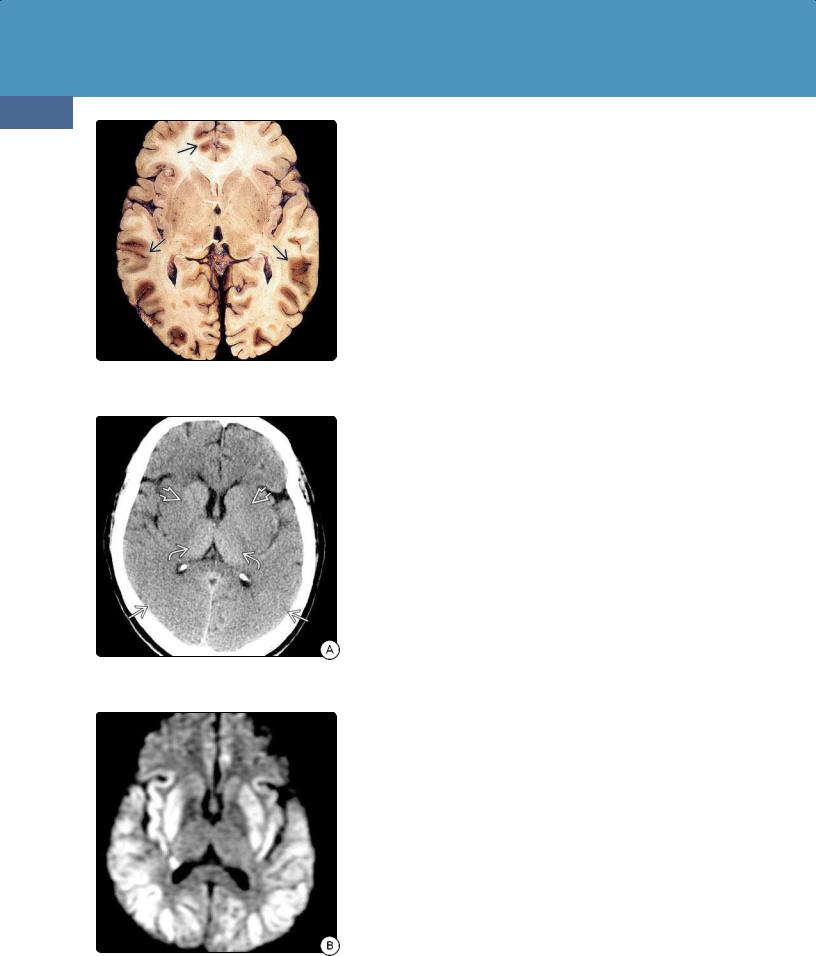
(32-12) Autopsy of severe hypoglycemia shows bilateral symmetric parietooccipital, frontal cortical necrosis . (Courtesy R. Hewlett, MD.)
(32-13A) NECT shows typical changes of hypoglycemia with parietooccipital gyral swelling, putamen hypodensity , spared thalami .
(32-13B) DWI in the same case of typical AHE shows restricted diffusion in parietooccipital cortex, putamina with thalamic and WM sparing.
and degree of hypoxia or other metabolic disturbances, cerebral blood flow, and CNS/cardiovascular metabolic requirements.
Pathology
Hypoglycemia has both direct and indirect effects on the brain, which is exquisitely sensitive to glucose insufficiency. Glucose insufficiency results in impaired oxygen utilization and compromised intracellular energy production. In addition, sustained glucose deprivation sensitizes microglial release of inflammatory mediators and may contribute to a number of comorbidities in diabetes and/or metabolic disorders.
Indirect effects of hypoglycemia occur secondary to the accumulation and release of excitatory neurotransmitters, which in turn accentuates the degree of hypoglycemic brain injury.
Cortical necrosis is the most common gross finding in hypoglycemic encephalopathy. Although the entire cortical ribbon can be affected, the parietooccipital regions are usually the most severely involved (32-12). Other especially vulnerable areas include the basal ganglia, hippocampi, and amygdalae. The thalami, white matter, brainstem, and cerebellum are typically spared.
Clinical Issues
The typical hypoglycemic patient is an elderly diabetic on insulin replacement therapy with altered dietary glucose intake. Deliberate or accidental insulin overdose is more common in children and young or middle-aged adults.
Seizures, mental status changes, and coma are common symptoms of hypoglycemic encephalopathy. Prognosis varies with the extent of brain injury. If the basal ganglia are involved, the outcome is generally poor. If basal ganglia injury is minimal or absent, residual neurologic deficits are correlated with the severity of cortical injury.
Because of sympathoadrenergic effects, myocardial infarction and severe arrhythmias are common indirect effects of hypoglycemia and may account for the "dead in bed" syndrome.
Imaging
CT Findings. NECT scans typically show symmetrically hypodense parietal and occipital lobes. The putamina frequently appear hypodense, whereas the thalami are spared (32-13). In severe cases, diffuse cerebral edema with near-total sulcal effacement and blurred gray-white matter interfaces can be seen.
MR Findings. T1 scans in patients with acute hypoglycemic encephalopathy can appear normal or demonstrate only gyral swelling and sulcal effacement. In the subacute and chronic stages, curvilinear gyral hyperintensity secondary to laminar necrosis may be present.
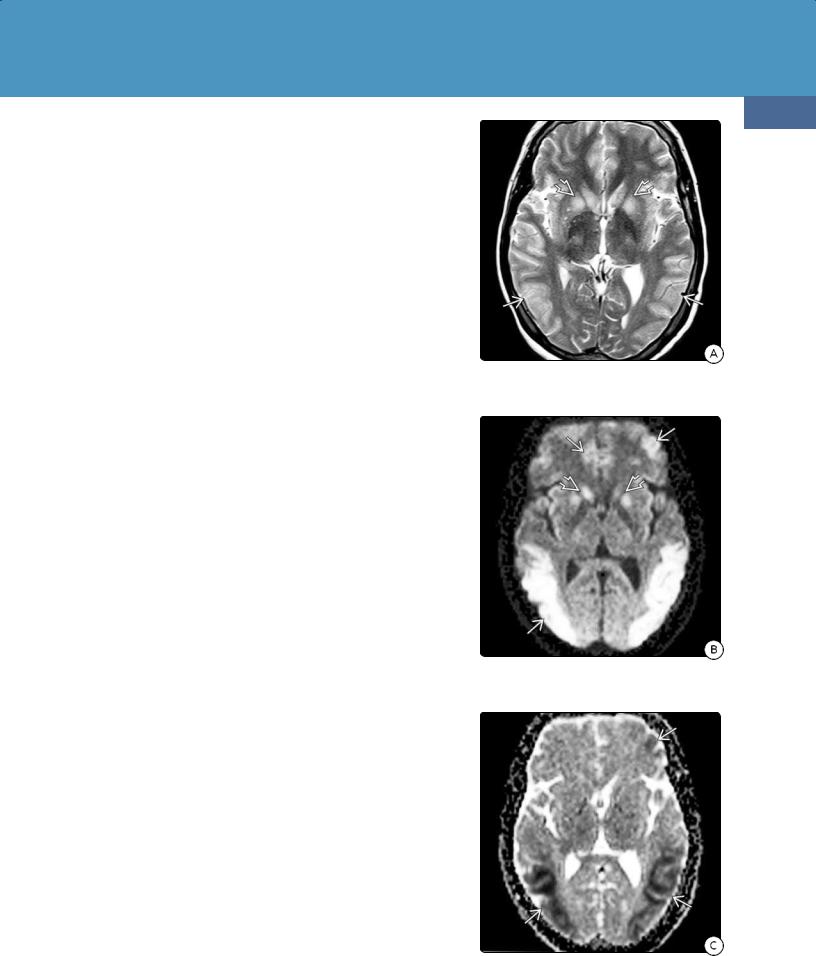
T2/FLAIR hyperintensity in the parietooccipital cortex and basal ganglia is typical of acute hypoglycemic encephalopathy (32-14). The thalami, subcortical/deep WM, and cerebellum are generally spared. T2* scans generally show minimal or no "blooming" to suggest hemorrhage. Enhancement on T1 C+ is variable and, when present, usually mild.
DWI scans show restricted diffusion in the affected areas, predominately the posterior parietal and occipital cortex (32-14). Cytotoxic corpus callosum splenium lesions with restricted diffusion have also been reported in association with hypoglycemia.
MRS demonstrates reduced NAA with or without a prominent lactate peak.
Acquired Metabolic and Systemic Disorders
Differential Diagnosis
The most important differential diagnosis of hypoglycemic encephalopathy is hypoxic-ischemic encephalopathy (HIE). HIE typically occurs following cardiac arrest or global hypoperfusion. In contrast to hypoglycemic encephalopathy, the thalami and cerebellum are often affected in HIE.
Acute cerebral ischemia-infarction is wedge-shaped, involving both the cortex and underlying WM.
Acute hypertensive encephalopathy (PRES) typically affects the parietooccipital cortex but spares most of the underlying WM and rarely restricts on DWI. PRES patients present with uncontrolled hypertension, not hypoglycemia.
Neonatal/Infantile Hypoglycemia
The immature brain is relatively resistant to hypoglycemia. Unlike older children and adults, neonates have lower absolute glucose demands and can utilize other substrates such as lactate to produce energy. Nevertheless, prolonged and/or severe hypoglycemia can result in devastating brain injury in newborn infants.
Terminology
The precise clinical/laboratory definition of neonatal/infantile hypoglycemia is controversial. Between 5 and 15% of normal term infants have initial plasma glucose values as low as 40-45 mg/dL. Currently accepted definitions of significant hypoglycemia in the newborn are glucose levels below 30-35 mg/dL in the first 24 hours after birth and 40-45 mg/dL thereafter.
Etiology
Congenital hyperinsulinism (HI) is the most common, most severe cause of persistent hypoglycemia in neonates and children. Mutations in 11 different genes cause congenital HI. The most common are inactivating mutations in the genes that encode the pancreatic β-cell ATP-sensitive potassium (KATP) channel.
Neonatal/infantile hypoglycemic encephalopathy is most often caused by maternal diabetes with poor glycemic control. Uncontrolled maternal diabetes leads to chronic fetal hyperglycemia in utero. This results in transient neonatal hyperinsulinemia and hypoglycemia of varying severity.
Neonatal hypoglycemia may also occur in the setting of BeckwithWiedemann syndrome (BWS), an inherited disorder with macrosomia, macroglossia, visceromegaly, omphalocele, embryonal tumors, adrenocortical cytomegaly, and renal abnormalities. Hypoglycemia due to HI occurs in approximately 50% of BWS cases. In most cases, BWS-associated hypoglycemia is mild and transient but can persist and—if undetected and untreated—pose significant risk for developmental sequelae.
Pathology
Transient, mild hypoglycemia generally does not injure the neonatal brain. Prolonged, severe hypoglycemia causes coagulative necrosis in the middle layers of the parietooccipital cortex and underlying WM.
Clinical Issues
Neonatal/infantile hypoglycemic encephalopathy typically presents in the first 3 days of life, usually within the first 24 hours. Large for gestation age babies have an increased risk of hypoglycemia even when they are not the product of diabetic pregnancies.
1031
(32-14A) T2WI in a 21y diabetic patient "found down" shows bilateral parietooccipital corticaland basal ganglia hyperintensity .
(32-14B) DWI in the same patient shows diffusion restriction in the cortex and basal ganglia . The thalami and WM are spared.
(32-14C) ADC map confirms restriction in the cortex .
Toxic, Metabolic, Degenerative, and CSF Disorders
1032
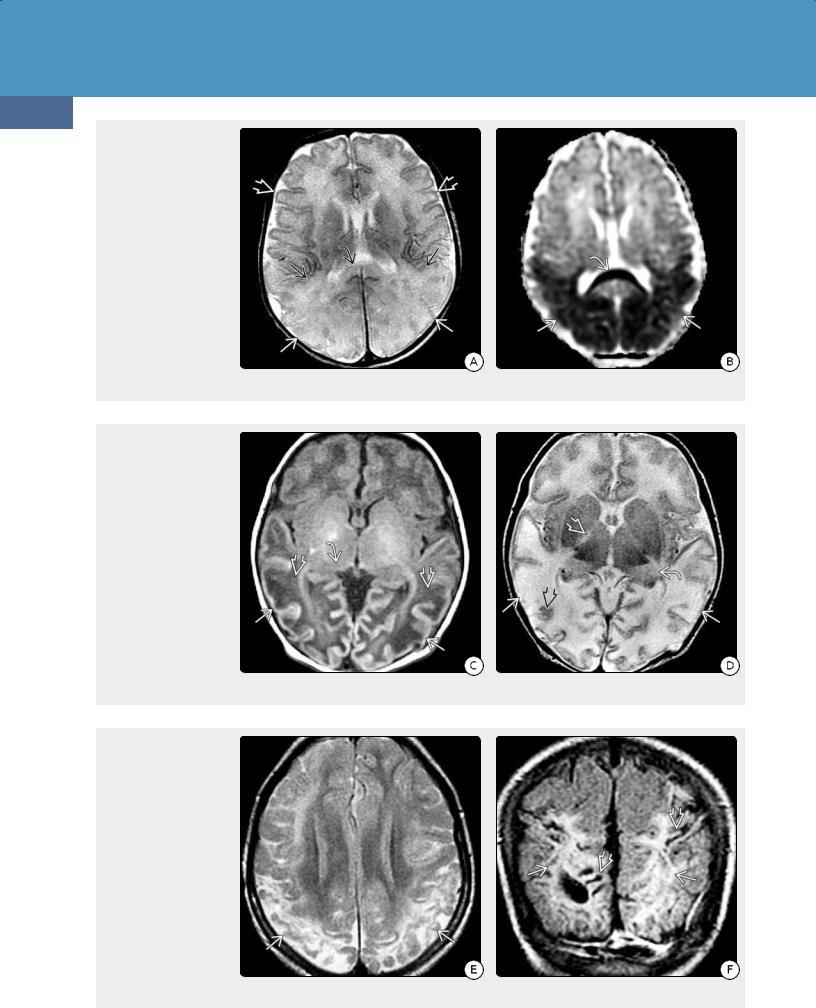
(32-15A) Axial T2WI in a
5d infant with hypoglycemia shows effaced GM-WM interfaces in the parietooccipital lobes compared with the normal frontal lobes . The cortex, underlying WM , and corpus callosum splenium are swollen and hyperintense. (3215B) ADC map in the same patient shows profound restricted diffusion in the parietal and occipital lobes and corpus callosum splenium .
(32-15C) T1WI in the same patient at 7 days shows swollen, markedly hypointense WM in the parietal and occipital lobes , hyperintense, thinned cortex , and hypointense pulvinars of the thalami . (32-15D) T2WI at the same time shows thinned swollen cortex with patchy increased and decreased signal intensity, hyperintense pulvinars , and abnormally hyperintense internal capsules .
(32-15E) Axial T2WI in the same infant at 1 year of age shows shrunken, hyperintense parietooccipital lobes with profound cortical loss, ulegyria, and encephalomalacicappearing WM . (3215F) Coronal FLAIR scan, also performed at 1 year of age, shows extensive WM hyperintensity and thinned, shrunken gyri with focally enlarged sulci.

Acquired Metabolic and Systemic Disorders
The precise level of hypoglycemia that requires treatment is controversial. Some experts recommend treating only symptomatic neonates with glucose concentrations below 4550 mg/dL. The response to glucose therapy is typically prompt if the degree and duration of hypoglycemia are mild to moderate.
Hypoglycemia remains an important cause of brain injury in children with HI. Up to 50% suffer from neurodevelopmental disabilities.
HYPOGLYCEMIA
General Concepts
•Imbalance between glucose supply, utilization → hypoglycemia
•Can be mild, transient, asymptomatic
•Extent of brain injury depends on
○Degree, duration of hypoglycemia
○CBF, glucose utilization
○Availability/utilization of alternative energy sources (e.g., lactate)
○Exacerbating factors (e.g., hypoxia)
○Recognition, prompt/appropriate treatment
Pediatric/Adult Hypoglycemia
•Etiology
○Usually associated with diabetes
○Absolute/relative insulin excess or glucose insufficiency
○Energy production/O utilization ↓, excitotoxic neurotransmitters ↑
•Pathology
○Cortical necrosis
•Imaging
○Hypodense/hyperintense parietooccipital cortex, basal ganglia
○WM, thalami, cerebellum generally spared
○Restricted diffusion common
○May cause reversible corpus callosum splenium lesion
Neonatal/Infantile Hypoglycemia
•Etiology
○Most common cause of transient hypoglycemia = maternal diabetes
○Fetal hyperglycemia → neonatal hyperinsulinemia → hypoglycemia
○Most common cause of severe, persistent hypoglycemia = congenital hyperinsulinemia (KATP mutation in 60%)
•Clinical issues
○Usually presents in first 3 postnatal days
○Glucose levels variable
•Imaging
○Often similar to adult (posterior predominance)
○Different: subcortical WM, thalami often involved
•Differential diagnosis
○Term hypoxic-ischemic encephalopathy
○Mitochondrial encephalopathy (MELAS)
1033
Imaging
Some imaging findings in neonatal/infantile hypoglycemia resemble those of older children and adults, i.e., predominant involvement of the parietooccipital cortex and basal ganglia. However, white matter, thalamic, and cerebellar involvement are all relatively more common in neonates compared with hypoglycemic encephalopathy in older children and adults.
NECT scans in neonates with acute hypoglycemic encephalopathy show posterior brain hypodensity with effaced gray-white matter interfaces. In especially severe cases, the brain appears diffusely swollen and hypodense.
MR scans in the acute stages of neonatal hypoglycemic encephalopathy show T2/FLAIR hyperintensity and restricted diffusion in the parietooccipital cortex, subcortical WM, and corpus callosum splenium (32-15A) (32-15B).
In the late acute/early subacute phase, the affected areas are swollen and edematous (32-15C) (32-15D).
Cystic encephalomalacia may ensue. In chronic hypoglycemic encephalopathy, the parietooccipital cortices become atrophic, shrunken, and encephalomalacic (32-15E) (32-15F).
Differential Diagnosis
As with older children and adults, the major differential diagnosis of neonatal hypoglycemic encephalopathy is term hypoxic-ischemic injury (HII). Hypoglycemic encephalopathy and HII often coexist, potentiating the extent of brain injury. Imaging findings in the two disorders may be indistinguishable.
Inherited mitochondrial disorders such as mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may present with cortical swelling that spares the underlying WM. MELAS is rarely bilaterally symmetric and demonstrates much more markedly elevated lactate on MRS.
Hyperglycemia-Associated Disorders
There are three serious complications of diabetes mellitus (DM) that require prompt recognition, diagnosis, and treatment: (1) hypoglycemia (see above), (2) diabetic ketoacidosis (DKA), and (3) hyperglycemic hyperosmolar state (HHS).
In the past, there has been relatively little concern about the influence of hyperglycemia on the CNS. The accepted clinical axiom has been that, in diabetics, it is better to err on the side of having high blood glucose because of the greater risk of brain and physical injury associated with severe hypoglycemia. After all, the conventional thinking went the brain is protected from high glucose exposure by the blood-brain barrier, which reduces brain exposure to two-thirds that of the peripheral blood level.
Accumulating evidence now shows that hyperglycemia is toxic for the brain at any and all stages of life. In this section, we briefly discuss DM itself before turning our attention specifically to hyperglycemia-induced brain injury. We first
Toxic, Metabolic, Degenerative, and CSF Disorders
1034
discuss the effects of chronic hyperglycemia on the brain, primarily seen as accelerated small vessel (microvascular) disease. We then consider two less common conditions associated with acute hyperglycemic changes in the CNS: DKA and HHS.
Diabetes
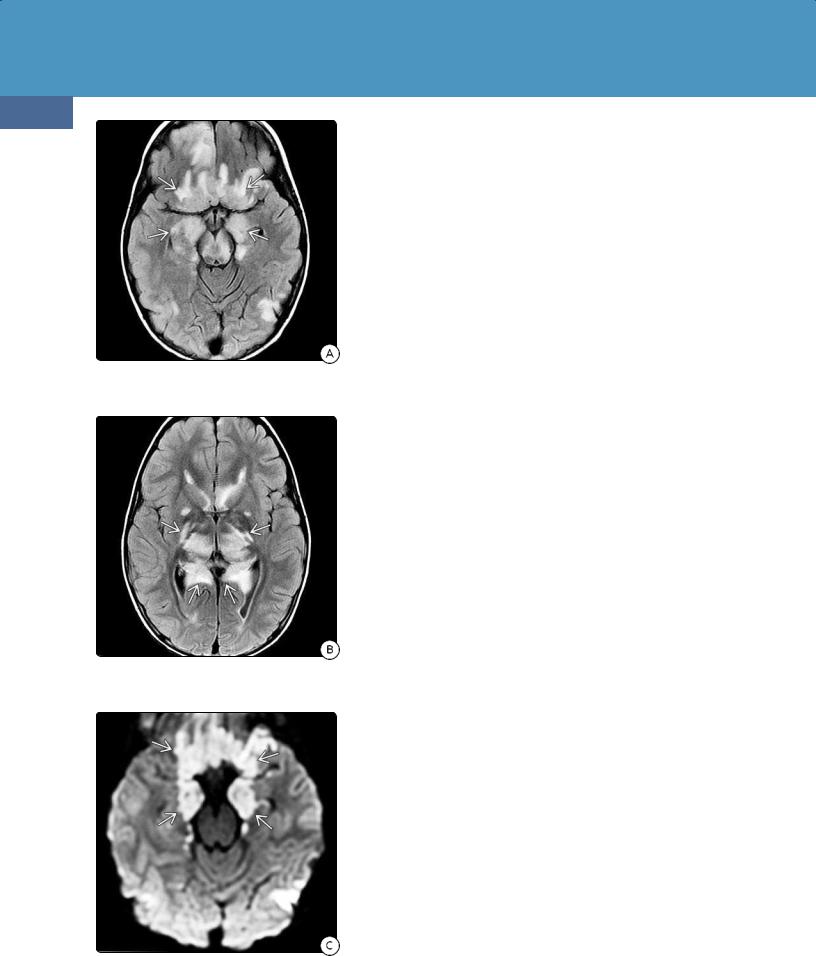
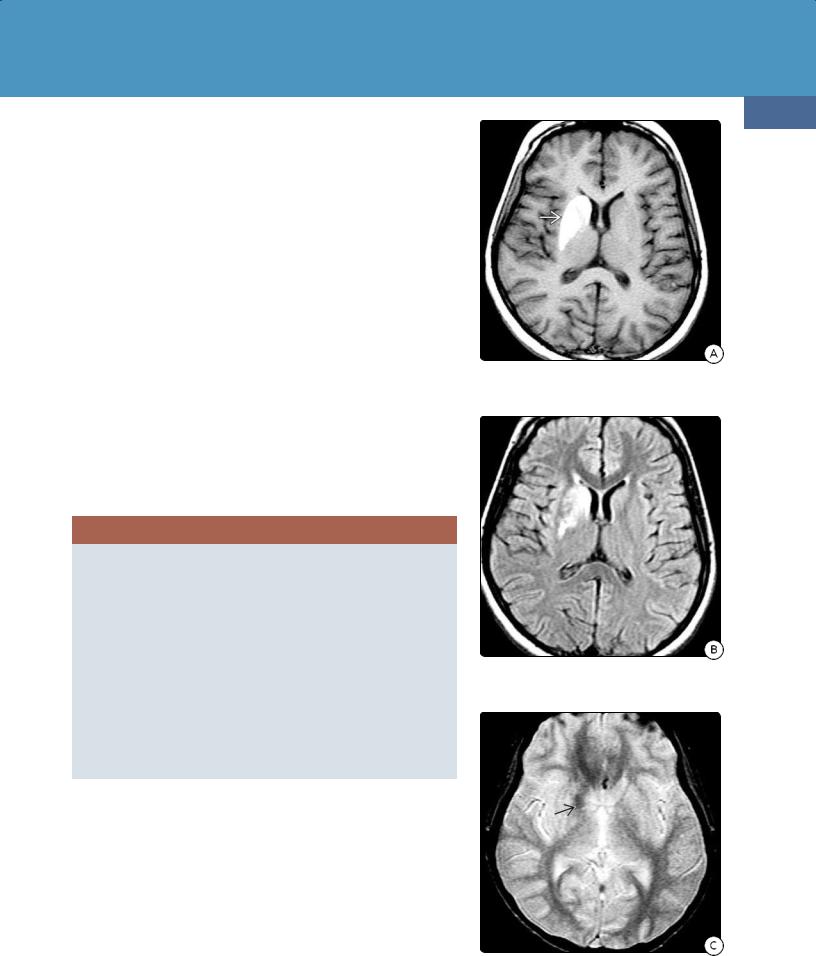
(32-16A) FLAIR in a child treated for DKAassociated cerebral edema shows hyperintensityfrom descending transtentorial herniation.
(32-16B) More cephalad FLAIR shows additional central hyperintensity .
(32-16C) DWI shows restricted diffusion from bilateral central descending transtentorial herniation. Compare with Figure 3-10.
In type 1 diabetes (DM1), previously known as "juvenile diabetes," a lack of insulin results from the destruction of insulin-producing β cells in the pancreas, presumably secondary to an autoimmune-mediated process. DM1 accounts for only 5-10% of all patients with DM. Children with DM1 have slower growth of gray and white matter during the period of rapid brain maturation. Metaanalytic reviews have also documented subtle but real neurocognitive deficits in both children and adults with DM1.
The vast majority of patients with diabetes have type 2 diabetes (DM2), previously termed "adult-onset diabetes." DM2 is also known as non-insulin- dependent diabetes and is caused by relative insulin deficiency or cellular insulin resistance. DM2 occurs in both children and adults. Risk factors include low activity level, poor diet, and excess body weight.
Chronic Hyperglycemic Brain Injury
Hyperglycemia-induced brain injury can be chronic or acute. With the worldwide rise in obesity and the soaring prevalence of DM2, the effects of chronic hyperglycemia on the brain are increasingly recognized. Patients with DM2 have accelerated arteriolosclerosis and lipohyalinosis with silent infarcts, brain volume loss, and decreased cognitive functioning.
MR shows increased numbers of T2/FLAIR subcortical and periventricular hyperintensities, especially in the frontal WM, pons, and cerebellum. DTI demonstrates loss of microstructural integrity with decreased FA. Elevated myoinositol on MRS reflects gliosis, an indicator of brain injury.
Acute Hyperglycemic Brain Injury
Although acute brain injury in hyperglycemia is less common than in hypoglycemia, hyperglycemia can also cause major morbidity and significant mortality. Two acute conditions are associated with hyperglycemia: DKA and HHS. These two diseases can be considered the endpoints of a clinical laboratory continuum from DKA with minimal symptoms and normal osmolality to HHS with minimal or no ketosis and coma.
DKA and HHS are both caused by reduction in the net effective action of circulating insulin. Intracellular starvation stimulates the release of the counterregulatory hormones, glucagon, catecholamines, cortisol, and growth hormones. This leads to accelerated hepatic and renal glucose production and impaired glucose utilization in insulin-dependent peripheral tissues (e.g., muscle, liver, and adipose). The result is hyperglycemia, lipolysis (with release of free fatty acids into the circulation), and hepatic fatty acid oxidation (to ketone bodies).
DKA and HHS are also associated with glycosuria, which can cause osmotic diuresis with subsequent loss of water, sodium, potassium, and other electrolytes. In severe cases, secondary changes of acute osmotic demyelination can complicate the imaging findings of both disorders.
Diabetic Ketoacidosis. Although DKA can occur in patients with both DM1 and DM2, it is quite uncommon in DM2. DKA is defined as acidosis with a venous pH less than 7.3 or serum bicarbonate concentration less than 15 mmol/L in the presence of serum glucose concentration more than 11 mmol/L. DKA is characterized by glucosuria, ketonuria, and ketonemia.
Acquired Metabolic and Systemic Disorders
DKA is often a recurrent disease. Mortality for each episode is relatively small (0.15-0.30%). Idiopathic cerebral edema accounts for at least two-thirds of fatal cases.
Imaging in patients with acute DKA is nonspecific, with vasogenic cerebral edema the most common abnormality. Younger age, severe acidosis, hypocapnia, and dehydration have all been cited as risk factors for DKArelated cerebral edema. If severe cerebral edema develops and is aggressively treated, survivors may show imaging evidence for bilateral (central) descending transtentorial herniation (32-16).
Hyperglycemic Hyperosmolar State. HHS occurs almost exclusively in patients with DM2. Once considered a relatively rare condition seen only in the elderly population, the emergence of childhood DM2 means HHS now occurs in patients of all ages and is becoming significantly more common.
The clinical laboratory criteria for HHS include plasma glucose concentration > 33.3 mmol/L, serum bicarbonate concentration > 15 mmol/L, absent or minimal ketonuria and ketonemia, effective serum osmolality > 320 mOsm/kg, and the presence of stupor or coma. Unlike DKA, seizures are common. Glycosuria and hypernatremia from dehydration can lead to cerebral edema, osmotic demyelination, seizure, and cardiac arrest.
Imaging in HHS is uncommon. T2/FLAIR hypointensity in the parietooccipital WM and transient lesions of the corpus callosum splenium have been reported. Patients treated for HHS with rapid correction of the hyperosmolar state may also develop osmotic demyelination with typical findings of central pontine myelinolysis (see below).
HYPERGLYCEMIA
General Concepts
•Spontaneous (rare) or diabetes-associated (common)
•Acute (rare) or chronic (common)
Hyperglycemia
•Chronic diabetes-associated brain injury
○Accelerated arteriolosclerosis, impaired cognition
○MR: decreased brain volume, increased WM hyperintensities
•Acute hypoglycemic brain injury
○Diabetic ketoacidosis (DKA)
○Hyperglycemic hyperosmolar state (HHS)
○Hyperglycemia-induced hemichorea-hemiballismus (HIHH)
•Imaging
○DKA: vasogenic cerebral edema ± osmotic demyelination
○HHS: subcortical WM hypointensity
○HIHH: unilateral T1 shortening in basal ganglia
Hyperglycemia-Induced Hemichorea-Hemiballismus
Also called delayed-onset diabetic striatopathy, the syndrome of hyperglycemia-induced hemichorea-hemiballismus (HIHH) is characterized by nonpatterned and involuntary unilateral movements. HIHH is a rare but potentially reversible complication of nonketotic hyperglycemia.
HIHH usually occurs in elderly patients, more often affects female patients, and may be the first ("unmasking") symptom of DM2. It can present as an acute, life-threatening manifestation of DM or develop 1-4 weeks after the hyperglycemic event when blood sugar has been controlled. Symptoms may resolve over days or persist for years.
MR findings are virtually pathognomonic of HIHH. T1 shortening in the basal ganglia contralateral to the hemichorea-hemiballism with sparing of the
1035
(32-17A) T1WI in hyperglycemia-induced hemichorea-hemiballismus (HIHH) shows uniformly hyperintense right basal ganglia .
(32-17B) FLAIR in the same patient shows patchy hyperintensity in the right basal ganglia. The remainder of the brain appears normal.
(32-17C) T2* GRE demonstrates minimal hypointensity in the globus pallidus . (Courtesy K. K. Oguz, MD.)